
来源:CBG资讯
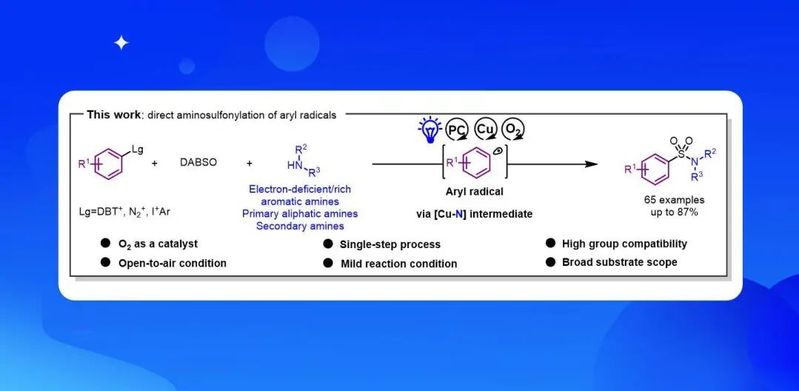
导语
尽管芳基磺酰胺类化合物在当代农药和医药领域中频繁出现,但其合成依然受限于预制备的有机硫化物、底物的亲核性和官能团的兼容性。针对以上问题,南开大学汪清民教授课题组在这一领域取得突破。
相关研究成果发表于ACS Catalysis(DOI: 10.1021/acscatal.3c03096)。
作者通过光氧化还原和铜协同催化,在室温和空气条件下,实现了从芳基自由基前体(芳基硫鎓盐、芳基重氮盐和芳基碘鎓)到芳基磺酰胺的一步法高效合成。反应直接使用商业易得的胺作片段,特别是对一些亲核性弱的胺,也有很好的适用性。机理研究显示,空气中的氧气对于两种催化循环是至关重要的,起到催化的作用。
前沿科研成果
光氧化还原与铜协同催化合成芳基磺酰胺
自从第一个磺酰胺类抗菌药物——Prontosil于1932年被发现,临床相关的合成芳基磺酰胺类药物便频繁出现,被用作抗生素、利尿剂、抗癌药物和抗精神病药物等(图1A)。芳基磺酰胺具有独特的理化性质、化学和代谢稳定、含有多种的杂原子,以上特点相结合,可提高这类药物的疗效和生物利用率。芳基磺酰胺的传统合成方法是通过胺与磺酰氯亲核取代反应。然而,该方法受限于磺酰氯的稳定性和可用性、胺的亲核性、反应过程中官能团兼容性。其中,官能团化磺酰氯的制备需要芳烃的亲电氯磺酰化或芳基有机硫化合物的氧化氯化。这些过程依然暴露出官能团兼容性差且存在位点选择性、强酸性反应条件和难闻气味的硫醇作底物等问题。在许多芳基磺酰胺活性分子的合成中,当底物是弱亲核性的胺或特殊官能团化的磺酰氯时,即便是在苛刻的反应条件下,也不能得到满意的收率。因此,发展一种以丰富、廉价、易得的试剂作底物,高效的合成方法是十分重要的。
近几十年来,有机硫化物转化为磺胺类化合物的报道屡见不鲜。其中,部分报道集中在通过有机金属试剂与二氧化硫的串联或过渡金属催化的芳基卤化物、芳基硼酸、芳基硫鎓盐等试剂的亚磺酸化合成芳基亚磺酸盐,再通过氧化氯化,以额外的两步反应合成芳基磺酰胺(图1B)。与此相比,通过在碳和氮片段间之间直接插入一分子二氧化硫合成磺酰胺,则更具吸引力和挑战性。一些巧妙的方法已被报道,但这些报道通常使用一些结构简单、预活化或非商业可得的氮片段——例如肼、NaN3、N-氯胺、亚硝基芳烃或硝基芳烃。然而,这些底物往往需要多步合成,并产生大量试剂的浪费。理想情况下,直接使用胺作片段,将更具商业价值。在此之前,一些课题组也做出了尝试。吴劼课题组通过N-羟基苯并三唑磺酸酯中间体,报道了一种芳基重氮盐、DABSO和富电子胺的三组分反应来合成芳基磺酰胺。Willis课题组以芳基硼酸、DABSO和富电子胺作底物,报道了一种高温条件、铜催化的Chan-Lam-type反应合成芳基磺酰胺(图1B)。尽管上述研究实现了胺的直接利用,但底物范围依然存在明显的局限性,特别是对一些亲核性弱的胺。
近年来,随着芳基自由基转化研究的发展,实现芳基自由基对芳基磺酰胺快速转化,将有助于化学结构的多样性和天然产物或药物的后期功能化研究。值得注意的是,吴劼课题组曾通过磺酰基自由基-氮自由基的交叉偶联反应成功合成了芳基磺酰肼,但其他类型的胺均不能获得对应的产物。这种结果是受自由基持续效应的限制。偶联的实现需要两种自由基具有不同的寿命并在相同频率生成。为克服这一问题,作者关注到铜作为一种低毒性、高储量的金属,被广泛用于有机合成中。受经典的铜催化的Ullmann-type和Chan-Lam偶联反应启发,作者设想了通过Cu(II)胺配合物中间体捕获磺酰基自由基,然后还原消除来合成芳基磺酰胺。
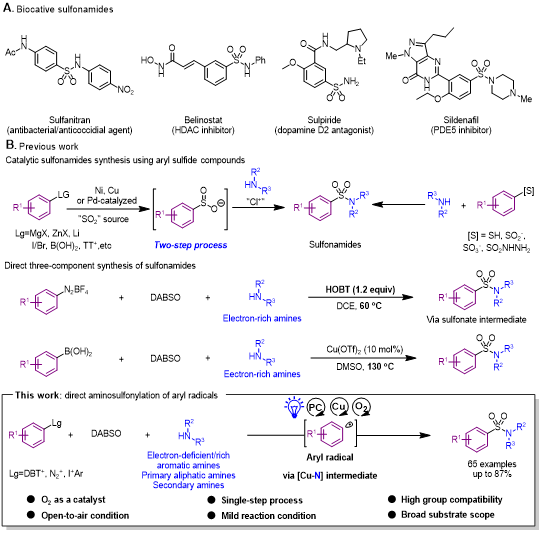
图1. 芳基磺酰胺相关的活性分子和研究背景(来源:ACS Catal.)
如表1所示,作者使用叔丁基苯制得二苯并噻吩硫鎓盐1a作芳基自由基前体,苯胺2a和1,4-二氮杂双环[2.2.2]辛烷-1,4-二鎓-1,4-二亚磺酸(DABSO, 1 equiv)作二氧化硫源,fac-Ir(ppy)3(2 mol%)作光催化剂,CuCl2(20 mol%)作金属催化剂,4,4-二叔丁基-2,2-联吡啶(dtbbpy, 20 mol%)作配体,吡啶(2 mol%)作碱,DCM作溶剂,在室温和空气条件下,用455nm蓝色LED灯照射24h,以85%分离产率得到目标产物3a(entry 1)。fac-Ir(ppy)3不能被10-苯基吩噻嗪代替(entry 2)。CuCl2不能被Cu(OAc)2代替(entry 2)。20 mol % CuCl2催化剂是最佳的(entry 4)。不加配体会导致收率大幅降低(entry 5)。控制实验表明:空气、光催化剂、铜催化剂和光是不可或缺(entries 6-10)。
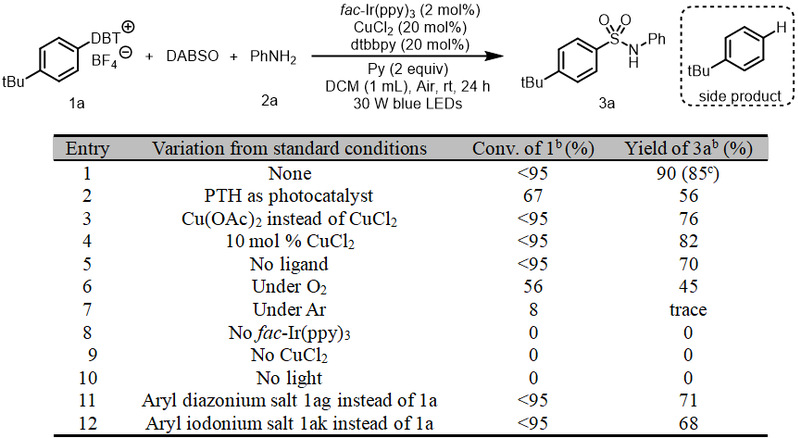
a Reaction conditions, unless otherwise noted: 1a (0.2 mmol), DABSO (0.2 mmol), 2a (0.4 mmol), fac-Ir(ppy)3 (2 mol %), CuCl2 (20 mol %), dtbbpy (20 mol %), and pyridine (Py, 0.4 mmol) in 1 mL of DCM were irradiated with 30 W blue LEDs (455 nm) at room temperature under air for 24 h. DBT = dibenzothiophenium, PTH = 10-phenyl-10H-phenothiazine. b Yields were determined by 1H NMR spectroscopy with mesitylene as an internal standard. c Isolated yield. 1ag = 4-(tert-butyl)benzenediazonium tetrafluoroborate; 1ak = (4-(tert-butyl)phenyl)(mesityl)iodonium triflate.
表1. 反应条件的筛选a(来源:ACS Catal.)
有了最佳反应条件后,作者对各种芳基硫鎓盐底物进行拓展(图2)。当苯基对位是环己基、4-甲基苯基或苯氧基时,反应分别以81%、82%和75%的产率得到产物3b、3c和3d。1-氯-3-苯基丙烷衍生盐1e以73%的产率得到产物3e。随后,作者对该反应进行克级制备,收率为59%。当底物是临,对位双取代的二甲苯衍生物时,产物3f的收率是63%。2,2-二氟-1,3-苯并二恶茂和1,4-苯并二恶烷衍生盐也适用于该反应,分别以58%和78%的收率得到相应产物3g和3h。含有多种卤素元素的底物均能以良好至中等的收率发生转化(3i-3l)。当底物中存在甲氧基和三氟甲氧基(3m)或甲酸酯基(3n)时,反应分别以55%和42%的产率得到产物。底物中含有酰胺和磺酰胺时,对应转化也能发生(3p,52%;3q,40%)。(S)-3-乙酰基-4-苯甲基-2-唑烷酮和2-甲氧基吡啶衍生盐以39%和48%产率得到相应的产物(3o和3r)。作者对一些天然产物和药物进行后期官能团化,以良好至中等的收率得到占吨酮衍生物3s、美西律衍生物3t、水杨甙衍生物3u、氟比洛芬衍生物3v、芬布芬衍生物3w、啶酰菌胺衍生物3x、噻奈普汀中间体衍生物3y、吡丙醚衍生物3z、吉非罗齐衍生物3aa和3ab。
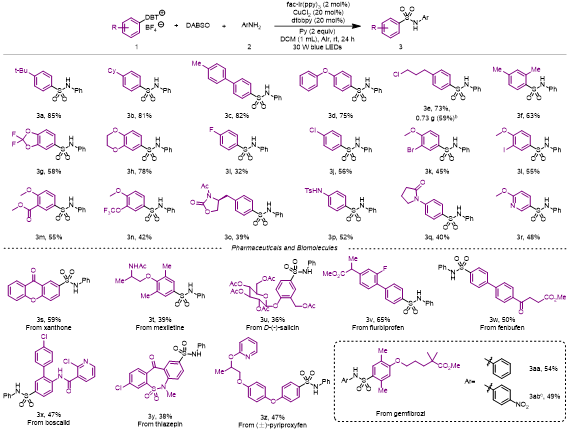
图2. 芳基硫蒽盐的底物拓展(来源:ACS Catal.)
随后,作者对胺的底物范围进行拓展(图3)。富电子苯胺——包括对甲氧基苯胺和3,5-二甲氧基苯胺,分别以59%和56%的产率得到产物4a和4o。当底物中含有一个或多个F、Cl、Br或I原子时,反应以56-74%得到产物4d-4h。值得注意的是,一些含有一个或两个拉电子基苯胺,包括4-三氟甲基苯胺、对硝基苯胺、间硝基苯胺、2,3,4,5,6-五氟苯胺、3-氰基苯胺、3-氨基-5-三氟甲基苯甲酸甲酯、3-氨基-5-氰基三氟甲苯、3-溴-4-硝基苯胺和3-氨基-4-甲基苯甲酸甲酯,均能以良好至中等的收率得到产物(4b, 4c, 4e, 4i-4n)。该方法同样适用于萘环、杂芳环和香豆素衍生胺,收率分别为61%(4p)、48%(4q)、67%(4r)和58%(4s)。虽然一级脂肪胺对铜催化剂有毒害作用,苄胺产物(4t)也以30%收率得到。作者通过进一步的条件筛选,使用Cu(OAc)2(20 mol%)和dofo(20 mol%)对仲胺底物进行拓展。N-甲基苯胺衍生物以45-78%的收率得到产物(4u-4w)。吲哚啉、吗啡啉、硫吗啡啉和哌嗪以28-70%的收率得到产物(4x-4ad)。

图3. 胺的底物拓展(来源:ACS Catal.)
作者进一步探索其他芳基自由基前体(图4A)。(4-(叔丁基)苯基)(均三甲苯基)碘鎓盐和4-叔丁基苯基重氮盐在该反应中的产率分别为72%、64%。2-甲基苯基重氮盐、3-溴苯基重氮盐和4-甲硫基苯基重氮盐均适用于该反应(5a, 41%;5b, 61%;5c, 47%)。苯并噻唑衍生物(5d)和香豆素衍生物(5e)以42%和69%的收率得到。合成sulfanitran传统的方法是以对硝基苯胺和对乙酰胺基苯磺酰氯作底物,需要吡啶作溶剂,收率仅为40%。然而,作者使用N-乙酰苯胺衍生盐(1ac)作底物,通过该反应,将收率提升至75%(图4B)。此外,作者以商业易得邻甲氧基苯甲酸为原料,经过三步,25%的总收率,合成了N-芳基-sulpiride衍生物(3ad)。
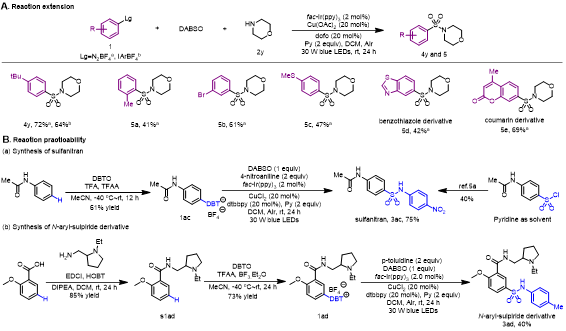
图4. 反应的应用(来源:ACS Catal.)
为了深入了解反应机理,作者进行了自由基捕获实验。发现加入TEMPO(2,2,6,6-四甲基胡椒酰氧基)或1,1’-二苯乙烯后,反应均被明显抑制。此外,通过高分辨率质谱也检测到自由基捕获产物6-8(图5a)。一系列荧光猝灭实验表明,激发态fac-Ir(ppy)3被1a猝灭最为明显(图5b)。这些结果证实:底物1a与激发态fac-Ir(ppy)3之间发生单电子转移。光开关实验表明,连续光照是产物生成的必要条件。量子产率(<1)也不支持自由基链过程。控制实验(表1,entry 7)显示,在氩气氛围中,反应被明显抑制。因此,作者排除了fac-IrIV(ppy)3+1氧化Cu(Ⅰ)配合物的可能性。薄层色谱和核磁氢谱均未观察到胺基亚磺酸中间体的生成。根据以往的报道,该物种被fac-IrIV(ppy)3+1氧化也是困难的。循环伏安法研究表明,DABSO作还原剂也可以被排除。
作者为了深入了解光催化循环,使用5,5-二甲基-1-吡咯啉-N-氧化物(DMPO)作为自由基捕获剂进行了电子顺磁共振研究(EPR,图5c)。硫鎓盐1a、苯胺2a、fac-Ir(ppy)3(2 mol%)和DMPO的组分在光照5 min后,显示出明显的信号。该信号归属于芳基自由基捕获加合物DMPO-1a(g = 2.002, AN = 14.23 g, AHb = 20.63 g)和DMPO氧化物(DMPOX; g = 2.002, AN = 14.06 g)。根据以往的经验,DMPOX是在光诱导氧化条件下生成的。作者在光催化体系中加入CuCl2(20 mol%)和dbbpy(20 mol%)后,观察到另一种信号。该信号归属于芳基自由基捕获加合物DMPO-1a(g = 2.002, AN = 14.43 g, AHb = 21.11 g)和羟基自由基捕获加合物DMPO-OH(g = 2.003, AN = 13.85 g, AHb = 14.94 g)。据以往的报道,在铜-氧还原反应中,O2可以作牺牲电子受体,转化为羟基自由基。这两组EPR实验显示,铜体系的加入可以抑制fac-IrIV(ppy)3+1氧化DMPO的过程,此外,也未观察到苯胺自由基阳离子捕获加合物。通过高分辨率质谱也没未检测到胺自由基偶联产物—偶氮苯。根据报道,在饱和甘汞电极(SCE)下,苯胺和4-碘苯胺的最大氧化电位约为0.90和0.94 V。作者在文章中使用到的一些缺电子胺,具有比苯胺更高的氧化电位。因此,fac-IrIV(ppy)3+1(E1/2IV/III = +0.77 V vs SCE)氧化胺,在热力学上至少高出0.13 V。在加入铜配合物后,DMPOX信号完全消失。作者推测:反应体系中生成的过氧铜复合物抑制了fac-IrIV(ppy)3+1氧化DMPO的过程。这些一系列结果显示,反应可能不涉及胺自由基的过程,并支持fac-IrIV(ppy)3+1氧化过氧铜复合物,回归基态光催化剂fac-Ir(ppy)3,完成光催化循环。
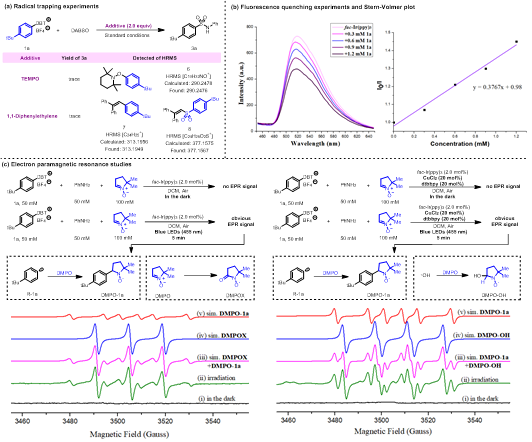
图5. 机理实验研究(来源:ACS Catal.)
根据上述机理研究和以往报道,作者提出了一种可能的反应机理(图6)。首先,激发态光催化剂* fac-Ir(ppy)3(E1/2IV/*III = -1.73V vs SCE)与自由基前体1发生单电子转移,得到还原态的1和fac-IrIV(ppy)3+1。还原态的1生成的芳基自由基被二氧化硫捕获,生成磺酰基自由基B。随后,B被铜(Ⅱ)胺复合物D捕获,生成铜(III)磺酰-氨复合物E。中间体E还原消除得到磺酰胺产物。生成的铜(Ⅰ)复合体F在碱性条件下,与胺的反应再次生成铜(Ⅰ)胺复合物C。空气中的氧气将铜(Ⅰ)胺复合物氧化为铜(Ⅱ)胺复合物D和铜(Ⅱ)过氧复合物G。最后,fac-IrIV(ppy)3+1 (E1/2IV/III = +0.77 V vs SCE)氧化中间体G,回归基态光催化剂fac-Ir(ppy)3,完成光催化循环。
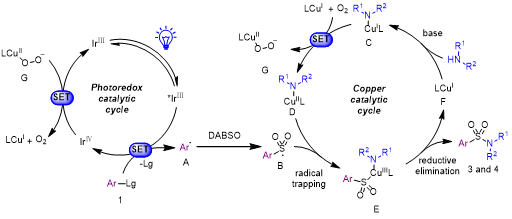
图6. 可能的反应机理(来源:ACS Catal.)
总结
作者通过光氧化还原和铜协同催化,实现了从芳基自由基前体(芳基硫鎓盐、芳基重氮盐和芳基碘鎓)到芳基磺酰胺的一步法高效合成。反应利用铜(Ⅱ)胺配合物捕获磺酰基自由基来克服自由基-自由基偶联的限制,同时具有良好的官能团兼容性,特别是对一些亲核弱的缺电子胺。通过该反应可以对于一些天然产物和药物进行后期磺酰胺化。机理研究支持氧气参与的催化过程。fac-IrIV(ppy)3+1氧化铜(Ⅱ)过氧复合物完成光催化循环。该方法实现了芳基自由基到芳基磺胺胺快速转化,有望促进相关化学生物学研究和药物发现。
本篇工作通讯作者为王兹稳教授和汪清民教授。南开大学博士研究生张铭君为该论文的第一作者。特别感谢杨茵特聘研究员在顺磁实验中的指导和帮助。上述研究工作得到了国家自然科学基金(21732002、22077071)和南开大学有机新物质创造前沿科学中心(63181206)的资助。
课题组简介

南开大学汪清民课题组隶属于南开大学元素有机化学国家重点实验室、有机新物质创造前沿科学中心和化学学院及天津化学化工协同创新中心。目前课题组拥有老师和研究生20多人。研究方向为生态农药、药物创制及环境友好的绿色合成反应。承担科技部、国家自然科学基金委、教育部、天津市等各种科研项目40多项。。先后在J. Agric. Food Chem.、Pest Manag. Sci.、Sci Adv.、Nat. Commun.、Angew. Chem. Int. Ed.、J. Med. Chem.、ACS.Catal、 Chem. Sci.、Green Chem.、ChemSusChem.、ACS Sustainable Chem. Eng.、Arthritis & Rheumatism、Org. Lett.、Chem. Commun.等杂志上发表论文300余篇。申请了100多项中国和美国及欧洲等发明专利,已授权80多项中国和美国及欧洲等发明专利。出版著作5部(章)。发明了仿生农药拟除虫菊酯系列产品和重多农药品种及高端精细化学品的清洁生产新方法,已成功应用于工业化大生产,产生了巨大的经济效益。创制了多个超高效的植物病毒病防治药剂和绿色杀虫杀螨剂候选品种以及国家Ⅰ类新药,处于产业化开发的不同阶段。培养毕业了30多名博士生和60多名硕士生,研究生获得了全国优秀博士学位论文提名奖、天津市优秀博士学位论文、南开大学优秀博士学位论文、南开大学优秀硕士学位论文和国家奖学金等。

关注“南开化学”微信公众号