
来源:电化学能源

能保持液态的电解质是确保可充电锂电池在宽温度范围内稳定运行并进行离子转移的最重要物理指标之一。一般认为,熔点高的强极性溶剂有利于电池在室温以上安全运行,但在低温(≤ -40℃)下容易结晶。
近日,南开大学陈军院士团队提出了一种结晶限制策略来解决这一问题。研究人员证明,尽管亚硫酸乙烯(ES,-17 ℃)和碳酸氟乙烯(FEC,~23 ℃)的熔点较高,但它们的混合物可以避免在低温下结晶,这可归因于低分子界面互作用和分子运动动力学的改变。合适的 ES/FEC 比率(10% FEC)可平衡离子的体传输和界面传输,从而使 LiNi0.8Mn0.1Co0.1O2||Li (NCM811||Li) 全电池在 -50 ℃ 至 +70 ℃ 的宽温度范围内具有出色的温度适应性和循环稳定性。与室温相比,该电池在 -50 ℃ 时的容量保持率超过 66%。NCM811||Li 软包电池在不同温度的实际条件下(电解液重量与正极容量比 (E/C)≤3.5 g Ah-1,负极与正极容量比 (N/P)≤1.09)表现出很高的循环稳定性。
【引言】
近几十年来,对更先进电池技术的无止境需求推动了全球对可充电锂金属电池(LMB)的重新探索。尽管在工程方面取得了长足进步,对 LMB 的科学理解也更加全面,但 LMB 在各种气候条件下(尤其是 ≤ -30℃ 和 ≥ 50℃)的可靠运行仍然是一项重大挑战。LMB对温度的适应能力较差的内在原因可归结为:1)低温下离子传输动力学不足,导致容量输出较低,并因枝晶突起的生长而产生严重的安全问题;2)高温下电解质和电极之间的寄生反应因反应性增加而加剧。原则上,所有这些问题都与电解质的化学性质密切相关。因此,开发在严酷温度下仍能可靠运行的先进电解质至关重要。
然而,大多数电解质要么无法覆盖全气候范围(无法在≤ -30℃或≥ 50℃的非典型温度下工作),要么需要特定设备(密封电池),这导致了重大的工程/技术挑战和能量密度损失。例如,以液化气或二乙醚为基础的电解质在低温电池中取得了巨大成功。然而,由于这些电解质的高挥发性和高蒸汽压,不借助特定设备在室温和高温下运行是不切实际的。
尽管 Fan 等人报道的全氟化电解质可实现耐高温 LMB,但由于成本和环境问题,其实际应用仍存在问题。对大多数溶剂来说,低熔点和高沸点在某种程度上是对立的,因为两者都受分子间作用力的支配。即使一个体系具有较宽的液态温度,也会影响其他所需的性能。与此同时,仅使用高沸点溶剂来开发全气候电解质的策略仍未开发出来,而这对提高电池的高温性能至关重要。因此,开发全气候电解质仍然面临着理论和材料可行性方面的挑战。
【要点】
在此,研究人员提出了一种开发全天候电解质的结晶限制策略。尽管亚硫酸乙烯(ES,-17 ℃)和碳酸氟乙烯(FEC,约 23 ℃)的熔点很高,但通过改变它们的比例,可以不断调整甚至规避 ES/FEC 混合电解质的冰点。
这种令人兴奋的现象可能是由于 ES/FEC 混合物的分子界面互作用减弱,取向排列比纯溶剂更加困难。此外,研究人员还发现,增加锂盐的不对称结构还能降低电解质的凝固点。在 ES/FEC 中含有 1M 高氯酸锂(LiClO4)的电解质可以在-60 ℃的严酷温度下稳定地传输离子。此外,适用的 ES/FEC 比率还有利于锂的电解质特性和界面化学性质,使锂沉积物形态致密,锂循环库仑效率(CE)高。优化后的电解液(1M LiClO4-ES/10% FEC)可使 NCM811||Li 全电池在 -50℃ 至 +70℃ 温度范围内稳定运行。此外,在实际条件下(N/P 比=1,E/C=3.5 g Ah-1),软包电池在较宽的温度范围内表现出较高的循环稳定性和出色的温度适应性。这项工作提供了一条将溶剂的高温和低温特性解耦的途径,拓宽了开发全气候电解质的溶剂范围。
【图文详情】
一、设计原理
根据热力学和动力学分析,研究人员利用混合溶剂策略来降低 ES/FEC 混合物的凝固点,而不会影响其他性能,从而开发出优异的全气候电解质。研究人员的设计原理如下:首先,从热力学角度来看,减少焓变和/或增加熵变可降低凝固点(等式 1)。一般来说,焓变由溶剂分子之间的分子界面互作用决定。减弱极性分子间的强相互作用原则上可以降低凝固焓,从而降低溶剂的凝固点。为此,研究人员选择了极性 S=O 基团比 C=O 基团弱的 ES 和高电负性氟的 FEC 来替代碳酸乙烯酯(EC)。另一方面,熵与分子的对称性有关。与对称分子相比,根据 Carnelley's 规则,不对称分子在冷冻过程中可能产生较小的焓变。与具有 C2 轴的 EC 分子相比,ES 和 FEC 只具有同一对称性(图 1a)。因此,与 ES/ES 和 FEC/FEC 混合物相比,不对称的 ES/FEC 混合物在冷冻过程中可能会产生更大的熵变。

其中,Tf、ΔHf 和 ΔSf 分别指凝固点、凝固焓和凝固熵。
其次,结晶经历了一个从无序液体到有序固体的分子重排过程,在这一过程中,分子旋转和迁移是分子排列以及引发成核和晶体生长的必要条件。由于混合物的分子扩散和重新定向速率可通过溶剂比例进行调节,因此合适的 ES/FEC 比例有望从动力学角度延长成核和晶体生长时间。
第三,ES 和 FEC 具有环状结构,加上独特的 S 和 F 元素,可确保生成稳定的固态电解质中界面(SEI),并具有足够的界面钝化和离子传输特性。此外,改变 ES/FEC 的体积比有可能改变电解质的溶剂化结构,进而影响离子传导和界面稳定性。在锂盐方面,高氯酸锂(LiClO4)因其解离能力强、对铝无电化学腐蚀、离子导电性好而被选为锂盐。
更重要的是,由于钝化双电层效应,LiClO4 还能使 ES 电解质的氧化电位高于其他锂盐。
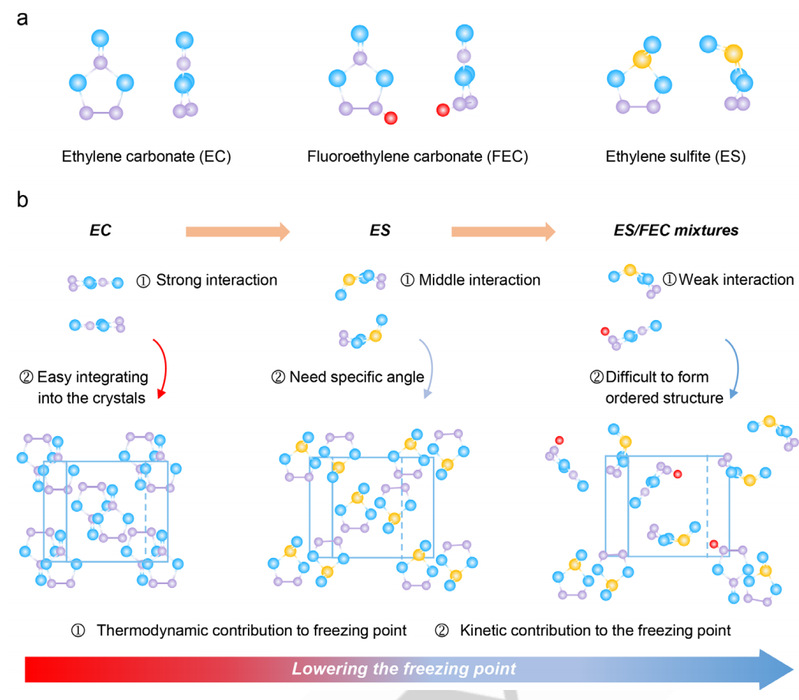
图 1.不对称溶剂全气候电解质的设计原理。(a) EC、FEC 和 ES 的分子结构。(b) 基于 ES/FEC 的电解质的设计原理示意图。与对称的电解质分子相比,不对称的 ES 分子由于活性面积较小,通常表现出较弱的分子相互作用。此外,非对称 ES 分子在冷冻过程中会比对称 ES 分子产生更显著的熵变。在凝固过程中,由于独特的排列角度,非对称 ES 分子可能比对称分子需要更长的时间。对称的电解质分子很容易融入晶体,而不对称的电解质分子往往会生成无序晶体,分子旋转需要额外的能量。对于不对称的 ES/FEC 混合物,由于溶剂的高熵升高和分子排列过程耗时,结晶变得更加困难。
二、分子界面互作用的减弱
静电势图显示了 ES 和 FEC 中原子的电荷密度。如图 2a 所示,ES 和 FEC 的高电子密度和低电子密度分别集中在 O 原子和 H 原子上,表明这些分子更有可能通过 O-H 键相互作用。此外,ES 的电子密度最低,呈平面外构型,电子密度低于 FEC,这意味着 ES 的分子相互作用弱于 FEC。此外,研究人员还对纯溶剂和混合溶剂(ES 或 FEC 被多个分子包裹)的分子相互作用进行了结构比较。如图 2b 所示,相互作用强度按 ES@FEC < FEC@ES < ES@ES < FEC@FEC 的顺序增加,这与上文讨论的设计规则十分吻合。此外,还考察了结晶状态下分子界面互作用与 FEC 含量的函数关系(图 2c)。随着 FEC 含量从 0% 增加到 70%,分子间的相互作用逐渐减弱。分子在液体和结晶处的相互作用较低,表明在热力学方面结晶的可能性较低。
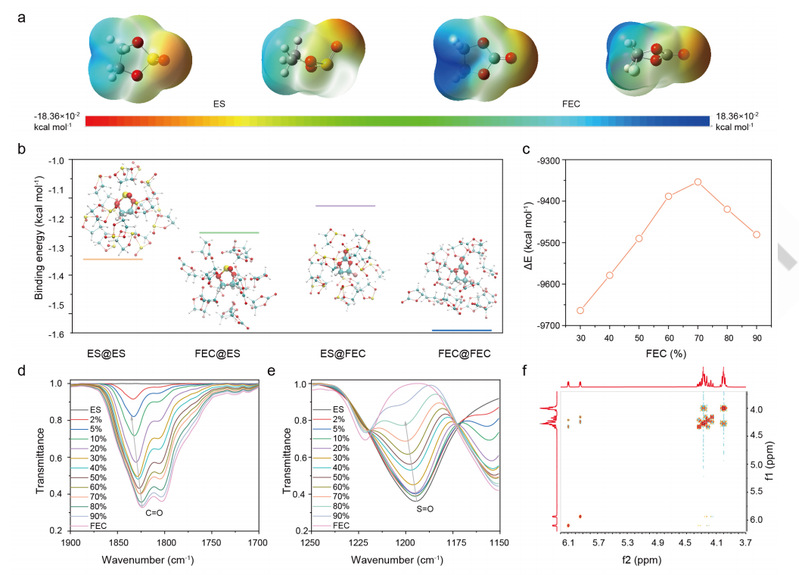
图 2.ES/FEC 混合物的热力学分析。
傅立叶变换红外图谱显示了 ES/FEC 混合物分子界面互作用的变化。如图 2d 所示,FEC 在 ~1823 cm-1 和 ~1800 cm-1 处显示出两个明显的峰值。
添加 ES 后,正如预期的那样,这些峰会向更高的波数移动(蓝移),并且随着 ES 含量的增加,更高的峰/更低的峰的相对强度也会增加。与纯 ES 相比,ES/FEC 混合物中 S=O 的伸缩振动也发生了蓝移(图 2e)。在拉曼图谱中也可以观察到类似的向高波长的移动。拉曼图谱和傅立叶变换红外图谱表明,FECES 的相互作用比 ES-ES 和 FEC-ES 的相互作用要弱得多,这与上述理论计算结果一致。此外,图 2f 中的NOESY表明 ES 和 FEC 的分子界面互作用很弱,因为与纯溶剂相比没有新的峰出现。
三、分子扩散和重新定向动力学
通过扩散有序核磁共振图谱(DOSY)分析了不同 ES/FEC 混合物的分子扩散动力学。
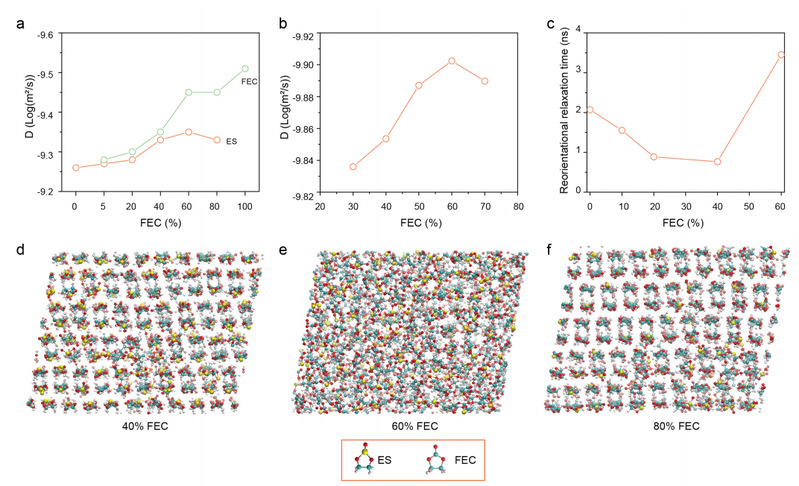
图 3.ES/FEC 混合物的动力学分析。
一般来说,分子移动速度越快,扩散系数就越高。如图 3a所示,纯 ES 的扩散比 FEC 的扩散远。然而,当 FEC 含量为 5%-40% 时,ES 和 FEC 在混合溶剂中的扩散系数几乎相同,这表明 ES 和 FEC 是以协调的方式受到限制和扩散的。当 FEC 含量较高时(>60%),ES 和 FEC 的扩散系数明显不同。虽然具体数值不同,但理论计算得出的 ES 扩散系数与实验结果显示出相同的趋势(图 3b)。
如图 3c 所示,ES/FEC 混合物的 τreor 首先随着 FEC 含量增加到 40% 而降低。τreor的降低意味着分子动力学速度更快。此外,根据 Adhikari 等人的研究结果,低 τreor 也表明分子间的相互作用较弱,这再次证实了图 2b、2f 中的结果。分子动力学模拟进一步证实了这一现象。在低温条件下,ES/FEC 混合物的形态从结晶态(40% FEC)变为无定形态(60% FEC),然后又变回结晶态(80% FEC)(图 3d-f )。因此,缓慢的分子扩散会延长结晶时间,而快速的重新定向弛豫时间则会降低分子融入晶体的概率,这两种情况都会降低 ES/FEC 混合物的凝固点。
四、溶剂和电解质的热物理性质
差示扫描量热法(DSC)测定了 ES/FEC 混合物的凝固点变化。如图 4a 所示,随着 FEC 含量从 0% 增加到 20%(ES/FEC 混合物中 FEC 的体积分数),混合物的凝固点从 -55.9℃ 骤降至 -71.7℃。计算得出的结晶焓显示出相同的趋势,证实了凝固点的热力学降低(图 4b)。此外,在 30% 至 70% 的 FEC 中,只能观察到一个与玻璃化转变温度 (Tg) 相对应的微小拐点(图 4a,插入部分)。一般来说,玻璃化转变意味着较差的凝固动力学,这表明需要一种动力学方法来降低凝固点。ES/FEC 混合物有限的凝固动力学也可以通过从 0% 到 30% 的 FEC 逐渐降低过冷温度来验证。
特别的,在 40% FEC 时,既观察不到冷晶体,也观察不到熔体,这与之前方法预测的二元相图不同。这些结果证实了研究人员通过具有不对称结构的溶剂降低凝固点的概念。
此外,还研究了 ES/EC 和四亚甲基砜 (SL)/EC 混合物,以验证溶剂不对称结构的重要性。ES/EC 和 SL/FEC 混合物与 ES/FEC 混合物在较低温度下的变化趋势相同。然而,在加热过程中,这些体系出现了冷晶体和熔体行为。这些结果表明,不对称结构在抑制混合物结晶方面是不可或缺的。值得注意的是,高 Tg 意味着溶剂运动动力学迟缓,会降低离子传输效率。因此,在后续研究中只选择了 FEC 含量小于 40% 的物质。
从原理上讲,Li+与偶极溶剂分子之间的强静电作用可以重新排列溶剂的配位结构,形成典型的 溶剂化结构;溶剂的原始偶极-偶极相互作用被部分破坏,转化为离子-偶极构型。因此,添加适当的盐可以延缓电解质溶剂的取向排列,从而降低凝固点。图 4c 显示了不同溶剂比的 1M LiClO4 ES/FEC 型电解质的 DSC 曲线。ES/FEC 型电解质的凝固峰从 FEC 的 0% 到 10% 之间向低温移动,然后完全消失(图 4c)。
此外,还发现锂盐的不对称结构也能降低电解质的凝固点。研究人员选择了三种具有代表性的酰亚胺盐:双(氟磺酰)酰亚胺锂(LiFSI)、双(三氟甲磺酰)酰亚胺锂(LiTFSI)和氟磺酰(三氟甲磺酰)酰亚胺锂(LiFTFSI)。1m LiFSI-ES 和 1m LiTFSI-ES 电解质的凝固点几乎相同,都高于 1M LiFTFSI-ES。因此,研究人员提出的通过不对称成分降低凝固点的概念可以扩展到盐类,尽管其详细机制尚未明确揭示,这还需要未来更多的工作。如上所述,考虑到电压窗口和降低冰点的能力,研究人员选择了氯化锂作为主要的锂盐。
此外,为了说明溶剂比例的影响,还进一步研究了具有代表性的电解质 1M LiClO4-ES、1M LiClO4-ES/10% FEC 和 1M LiClO4-ES/40% FEC。
电解液的低挥发性是确保电池在高温下安全运行的一个重要参数。如图 4d 所示,所有基于 ES/FEC 的电解液都具有出色的低挥发性,即使在 90℃ 时也能达到 90.4% 以上的质量保持率。可以合理地认为,随着 FEC 含量的增加,热稳定性也会提高。与此相反,对于商用电解质(1M LiPF6-EC/DEC 与 2% LiBOB,名称为 Com.),尽管其完全蒸发温度高于 ES/FEC 型电解质,但在 70℃ 之前就已观察到明显的质量下降,最高达 13.9%。这些结果保证了研究人员基于 ES/FEC 的电解质的高温运行。
五、电解质的性质和溶剂化结构
图 4e 显示了 ES/FEC 型电解质和 Com. 电解质的离子电导率测量值。所有电解质的电导率都随着温度的降低而逐渐降低。
在 0 ℃ 以上的温度下,所有电解质的离子导电率几乎相同。不过,在较低温度下可以观察到明显的区别。与此形成鲜明对比的是,所有基于 ES/FEC 的电解质在温度≥ -40 ℃ 时都保持液态,离子电导率超过 0.1 mS cm-1。当进一步降低操作温度时,1M LiClO4-ES 的离子电导率会在 -50 ℃ 时突然下降,而 1M LiClO4ES/10% FEC 则会在 -60 ℃ 时突然下降。相比之下,1M LiClO4-ES/40% FEC 即使在-70 ℃时也能保持良好的离子传输性能。
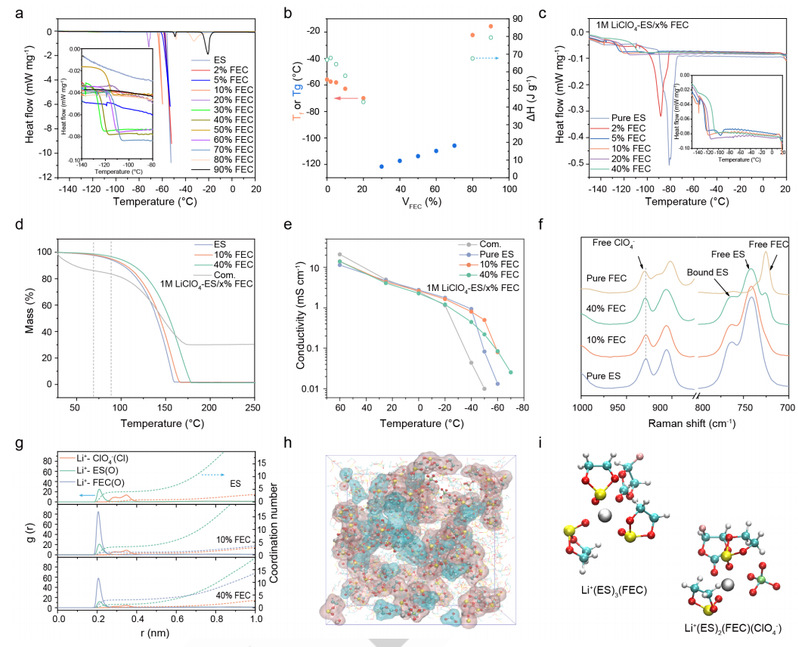
图 4.配制电解质的物理性质、电化学性能和溶剂化结构。
这一令人满意的结果表明,研究人员的混合溶剂策略可以有效降低溶剂的凝固点,同时允许离子在低温下快速传输。数据分析显示,1M LiClO4-ES/40% FEC 的离子电导率遵循 VTF 方程,而其他电解质则符合阿伦尼乌斯方程。
使用不锈钢电极作为工作电极进行线性扫描伏安(LSV)测量,以评估各种电解质的电化学稳定性窗口。商用电解质从 ~4.6 V 开始出现明显的阳极分解(根据 0.01 mA cm-2 时的阳极电流密度值确定)。此外,这个微小的峰值随着 FEC 的增加而增强,表明更多的 FEC 可能会恶化电极的钝化。进一步阳极扫描后,1M LiClO4-ES/10% FEC 在 4.9 V 之前没有检测到明显的氧化电流,高于 1M LiClO4-ES 的 4.7 V 和 1M LiClO4-ES/40% FEC 的 4.8 V。1M LiClO4-ES/10% FEC 在 4.7 V 之前的电流密度明显优于其他两种电解质。
为了了解 ES/FEC 型电解质氧化稳定性不同的原因,研究人员进行了拉曼图谱分析。如图 4f 所示,所有基于 ES/FEC 的电解质都显示出一种特征性的溶剂分离离子对(SSIP)结构,其中 Li+ 配位溶剂分子在第一溶剂化鞘中占主导地位。值得注意的是,随着 FEC 的增加,在 ~930 cm-1 处可以观察到微小的蓝移,这表明 ClO4 的配位减少了。为了进一步揭示每种电解质在原子水平上的局部环境,研究人员进行了 MD 模拟。
如图 4g 所示,径向分布函数 (RDF) 显示,FEC 与 Li+ 的结合比 ES 更紧密(FEC 为 2.06 Å,ES 为 2.12 Å),这意味着充电过程中阳极表面的 FEC 会优先减少。当 FEC 含量从 10% 增加到 40% 时,Li+-ES 的配位数 (CN) 从 3.87 下降到 3.07。此外,少量的 ClO4- 也参与了溶剂化结构。ClO4- 的 CN 随 FEC 含量的增加而降低,从 0.78(1M LiClO4-ES)降至 0.27(1M LiClO4-ES/40%FEC),与拉曼结果一致。
六、不同温度下的界面特性和锂沉积形态
在Li||Cu半电池中测试了基于 ES/FEC 的电解质的还原机制。加入 FEC 后,在还原 ES 之前又出现了一个还原峰。因此,界面化学性质是由 ES 和 FEC 的还原同时决定的。研究人员通过 X 射线光电子能谱 (XPS) 和深度剖面分析来研究基于 ES/FEC 的电解质中的 SEI 成分。考虑到 稀释 的盐浓度,SEI 主要来自溶剂分解的化合物,可通过 FEC 含量进行调整。研究发现,所有基于 ES/FEC 的电解质在使用 Ar+ 进行 40 纳米溅射时都能产生相似的 C 含量(图 5a-c)。添加 FEC 后,可观察到与 CO3 成分相对应的新峰值。
由 LiF 组成的 F 信号的出现表明了在 1M LiClO4-ES/10% FEC 中 FEC 溶剂的分解。锂原子浓度是代表 SEI 内部无机物比例的一个重要指标,它在 40% FEC 浓度时会降低。与室温相比,10% FEC 的 SEI 成分在 -40 ℃ 时保持相似的成分和含量(图 5d)。这些结果表明,与传统电解质在低温下产生的高度结晶和不均匀的 SEI 不同,ES/FEC 型电解质中被动 SEI 的形成对低温不敏感。
为了评估基于 ES/FEC 的电解质在不同温度下的离子传输效率,研究人员组装了Li||Li对称电池。电荷转移阻抗(Rct)在低温下主导着去溶剂化过程,随着温度的升高呈 V 形变化。在基于 ES/FEC 的电解质中,10% FEC 的 Rct 最低(图 5e)。另一方面,不同温度下电阻式 SEI(RSEI)内的裸 Li+ 传输与 Rct 的变化趋势相同,即在所有基于 ES/FEC 的电解质中,10% FEC 的 RSEI 最低(图 5f)。
1M LiClO4-ES/40% FEC 的离子导电电阻升高的原因是 SEI 成分中的无机物比率较低。然而,这并不能解释为什么 1M LiClO4-ES 的电阻比 1M LiClO4-ES/10% FEC 的电阻高,因为含 S 的无机成分具有很高的锂电导率。
根据以往的文献,SEI 溶解是 ES 型电解质的主要问题之一,会导致 SEI 不断破碎和重整;因此,ES 型电解质会伴随着快速电解质耗竭和低库仑效率(CE)。与 1M LiClO4-ES 在循环过程中检测到的严重 SEI 溶解不同,10% FEC 电解质在自制电池中循环五次后没有出现吸收峰或沉淀。因此,合适的 LiF 含量和稳定的界面是 1M LiClO4-ES/10% FEC 电解液离子导电电阻最低的原因。
为了了解锂沉积形态与电解质配方和界面化学成分之间的关系,研究人员采用了聚焦离子束扫描电子显微镜(FIB-SEM)。图 5g-r 显示了非对称Li||Cu电池的锂沉积形态与室温下不同 ES/FEC 比率的函数关系。对于纯 ES 而言,图 5g 中的锂沉积物是扁平的,具有相对较大的颗粒形态。
然而,从横截面观察可以发现许多空隙和少量针状锂沉积物。这种形态是由 SEI 的持续溶解和重塑造成的,原位紫外可见图谱和光学照片中的黄色沉积物都证实了这一点。相比之下,10% FEC 产生的锂沉积物更为致密,具有银白色光泽(图 5k )。此外,FIB-SEM 扫描仪检测的横截面形态进一步证实了所需的锂沉积物,观察到明显的紧密堆积锂形态。将 FEC 含量提高到 40% 并不能进一步改善锂沉积物的形态,因为 FEC 含量越高,Rct 和 RSEI 越大。因此,在 40% 的纤维素酯中观察到了带有大空隙的土丘状形态(图 5o)。
当工作温度降低到 -40℃ 时,锂沉积物的直径随着 FEC 含量的增加而直观地减小(图 5h、l、p)。研究人员强调,在低温条件下,纯 ES 电解质中 SEI 的溶解受到了很好的限制;因此,基于纯 ES 的电解质显示出致密的形态,没有明显的空隙(图 5h 和图 5i)。相比之下,由于离子传输受限,在 40% FEC 中会产生多孔和针状的锂沉积物(图 5p 和图 5q)。幸运的是,10% FEC 保持了大块的锂形态,具有均匀的全局覆盖和紧凑的堆叠,尽管其顶端部分比 ES 相对更加凹凸不平(图 5l 和图 5m)。
在 60 ℃ 时,1M LiClO4-ES 显示出粗糙多孔的锂沉积,并伴有大量裂纹和孔洞(图 5j)。这种形态可归因于 SEI 的严重溶解导致锂和电解质之间不断发生副反应。与此形成鲜明对比的是,在 1M LiClO4ES/10%FEC 中的锂沉积物在铜表面保持了金属光泽和无缝堆积的形态(图 5n)。令人惊讶的是,在 1M LiClO4-ES/40%FEC 中形成的锂沉积物也很光滑(图 5r)。根据上述结果,研究人员认为尽管 LiF 的离子传输特性较差,但它确实能提高 ES/FEC 型电解质的界面稳定性。因此,决定研究人员的电解质在不同温度下性能的主要因素是不同的。在低温条件下,本体电解质和界面层中迟缓的离子导电动力学对电化学性能起着主导作用。但在高温条件下,情况发生了变化,分子热运动的增强消除了离子传输的限制。因此,电化学性能取决于界面稳定性。因此,FEC 含量低有利于离子在低温下的传导(尤其是在本体电解质和界面电阻中),而 FEC 含量高则能增强界面稳定性。
使用基于 ES/FEC 的电解质的锂金属电池的电化学性能 Li||Cu半电池的 CE 验证了上述论点,即 FEC 含量较高的电解质可以降低熔点,但会影响离子转移和锂金属在低温下的可回收性,这一点从 1M LiClO4-ES/ 40% FEC 的较大过电位和相对较低的 CE 中可以看出。此外,使用 1M LiClO4-ES 的电池无法在 -50℃ 下工作,而使用 1M LiClO4-ES/10% FEC 的电池在相同温度下仍能保持较高的 CE。更重要的是,使用 10% FEC 的电池在 60℃ 时仍能保持较高的 CE 值,这表明它们在宽温条件下与锂金属阳极的兼容性很高,同时也证明了 SEI 良好的耐腐蚀性。1M LiClO4ES/10% FEC 性能均衡,可支持Li|Cu和Li|Li电池的稳定循环。研究人员还评估了基于 ES/FEC 的电解质应用于当代石墨负极的可行性。基于 ES/FEC 的电解质与石墨电极之间具有很高的电极兼容性。含有 10% FEC 的电解质允许 Li + 在石墨中进行可逆的插层和去插层,循环次数超过 500 次,室温下容量保持率为 76.9%。在较低温度(-20 ℃)下,1M LiClO4-ES/10% FEC 还支持石墨电极的出色可回收性,室温容量保持率为 84.2%。
受其全面性能的鼓舞,研究人员利用由金属锂阳极(50 μm)和商用 NCM811 阴极组成的全电池来评估 1M LiClO4-ES/10% FEC 的宽温适用性。图 6a 和 6b 展示了 NCM811||Li 全电池在 1M LiClO4-ES/10% FEC 和 Com. 电解质中不同温度下的放电电压曲线。在相同的 ~2 mAh cm-2 阴极负载条件下,两种电解质中的 NCM811||Li 全电池在 -10 ℃ 至 70 ℃ 的放电容量大致相同。然而,在低温条件下,放电容量和电压曲线出现明显差异。在 -20 ℃ 时,Com.电解质中的 NCM811||Li 全电池只能提供 78.6 mAh g-1 的低放电容量,且电压衰减明显。相比之下,使用 10% FEC 电解质的电池则显示出 165.1 mAh g-1 的高放电容量,且没有明显的过电位增加。
电化学阻抗谱(EIS)分析也表明,Com.电解质的 Rct 比基于 ES/FEC 的电解质大一个数量级。当温度降低到 -30 ℃ 时,Com. 电解质几乎无法支持电池的正常工作;只能实现 15.8 mAh g-1 的低容量。令人吃惊的是,使用 10% FEC 电解质的电池可以在 -50℃ 的超低温下存活,提供 139.8 mAh g-1 的高容量,与室温相比,容量保持率达到 66%。令人印象深刻的耐温性证实了研究人员基于 ES/FEC 的电解质的稳健性。
在实际应用中,电解液用量少的电池是实现高比能量的最关键参数之一。图 6c 显示了 NCM811||Li 全电池在室温下有限电解质(20 µL mAh-1)条件下的长期循环稳定性。研究人员基于 ES/FEC (10%) 的电解液可在 300 次循环后实现 ~91.8% 的容量保持率,平均 CE 高达 99.74%。
然而,Com.电解质仅在最初的 60 个循环中表现出稳定的循环,然后容量和 CE 迅速衰减。使用 Com. 电解质的电池突然失效可归因于电解质或锂耗竭。此外,在低温条件下也能观察到类似的循环趋势。
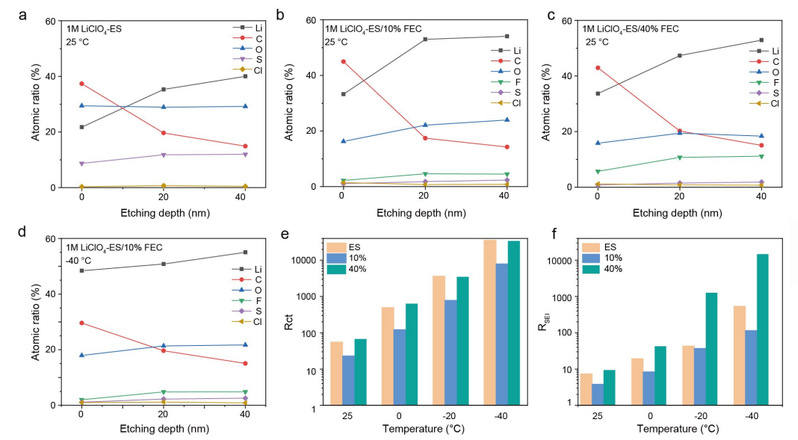

图 5 界面属性界面特性
如图 6d 所示,使用 Com. 电解质的电池在-20 ℃时仅显示出约 121.8 mAh g-1 的微不足道的容量;仅在 37 个循环后就迅速衰减。与此形成鲜明对比的是,使用 1M LiClO4-ES/10% FEC 的电池在-33 ℃时具有惊人的循环稳定性和高容量。即使在 -40 ℃ 的超低温条件下,1M LiClO4-ES/10% FEC 电解质也能使 NCM811||Li 全电池保持 130 mAh g-1 的高放电容量。值得强调的是,这些可靠的循环性能超过了最近报道的最先进的电解质。研究人员的 ES/FEC 电解质在高温条件下也具有出色的电化学性能。使用 1M LiClO4-ES/10% FEC 的全电池在 +50 ℃ 下可稳定工作 25 个循环。
这些出色的电化学性能促使研究人员在更现实的条件下进行进一步评估:更高的阴极负载(2.5 mAh cm-2)和较低的电解液量(8 µL mAh-1)。如图 6e 所示,采用 10% FEC 的 NCM811||Li 全电池在 250 次循环中保持了 80% 的容量,平均 CE 值高达 99.36%,令人印象深刻。根据现实条件的标准,研究人员进一步组装了锂和电解液用量有限的软包电池。如图 6f 所示,1M LiClO4ES/10% FEC 电解液可使小软包电池(约 0.87 Ah)稳定循环 140 多个周期,平均 CE 高达 99.5%。
此外,在第 1 次和第 20 次循环时,没有出现明显的过电位升高和电压下降,这表明电解质具有出色的稳定性,并与锂金属电池兼容。
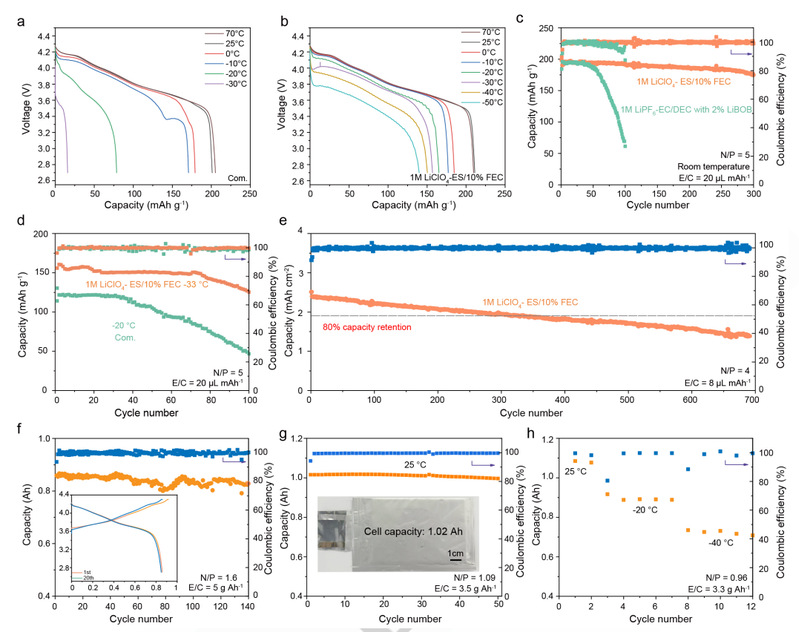
图 6.基于 ES/FEC 的电解质在不同温度下的电化学性能。
此外,当 N/P 和 E/C 比率分别进一步降低到 1.09 和 3.5 g Ah-1 时,NCM811||Li软包电池(1.02 Ah)在 50 次循环后的容量保持率为 97.7%(图 6g)。以前的许多研究只使用低容量的纽扣电池来评估电解质的宽温性能;这可能会导致高估耐温可逆性。如图 6h 所示,在 -20 ℃ 和 -40 ℃ 下,使用 1M LiClO4-ES/10% FEC 电解液的软包电池的相对容量分别比室温下保留了约 83.3% 和 68.7%。相应的电压曲线表明,软包电池在低温下的平均电压与纽扣电池相当,在 20 ℃ 和 -40 ℃ 时分别为 3.83 V 和 3.73 V。这些结果再次证明了研究人员基于 ES/FEC 的电解液具有良好的宽温性能。
【结论】
通过调节分子界面互作用和凝固动力学,研究人员成功地降低了具有不对称结构的强极性溶剂的凝固点,这显示了锂电池在低温下工作性能的优势。研究人员证明了 ES/FEC 比率不仅能调节电解质的凝固点,还能显著影响锂金属电池的电化学性能。高 FEC 含量(40%)有利于高温下的界面稳定性并支持电解质的低凝固点,而低 FEC 含量(低于 10%)则有利于低温下的离子传导。
利用这些见解,生产出了一种优化的全气候电解液,即 1M LiClO4-ES/10%FEC。使用这种电解液的 Li||NMC811 全电池可在 -50 至 +70 ℃ 的宽温度范围内提供出色的电化学性能。
更重要的是,研究人员证明了锂盐的不对称结构也能显著降低电解质的凝固点。这项研究强调了设计全气候液态电解质的一个关键但容易被忽视的方向,即非对称溶剂效应。研究人员的结晶限制策略提供了一条实用的途径,代表了实现宽使用温度范围的能源密集型锂金属电池的重大理论进展。
Asymmetric Solvents Regulated Crystallization-Limited Electrolytes for All-Climate Lithium Metal Batteries
Angewandte Chemie International Edition ( IF 16.6 ) Pub Date : 2023-12-15 , DOI: 10.1002/anie.202310905
Yuankun Wang, Zhiming Li, Weiwei Xie, Qiu Zhang, Zhenkun Hao, Chunyu Zheng, Jinze Hou, Yong Lu, Zhenhua Yan, Qing Zhao, Jun Chen

关注“南开化学”微信公众号