
来源:X-MOL
二氧化碳(CO2)作为一种理想的C1原料,其通过电合成技术转化为高附加值羧酸的前景备受关注。其中,苄位羧酸因广泛存在于布洛芬等药物及生物活性分子中而具有重要研究价值。目前,电化学苄位羧酸合成的研究主要集中在(伪)卤化物的羧化反应上,然而较低的原子经济性极大地限制了其实际应用价值(图1A)。相比之下,苄位C−H键的直接电化学羧化策略具有原子利用率高、经济性好的显著优势;然而苄位C−H键与二氧化碳固有的惰性所带来的挑战亟待解决。早期研究主要采用强碱促进的脱质子化/二氧化碳亲核进攻策略(图1B左)。近年来,光催化技术的利用不同光催化剂、过渡金属或氢原子转移试剂使得苄位C−H键的直接羧化成为可能(图1B右)。然而,针对一级、二级、三级C−H键与二氧化碳的通用羧化方法仍不完善,亟待进一步探索和发展。
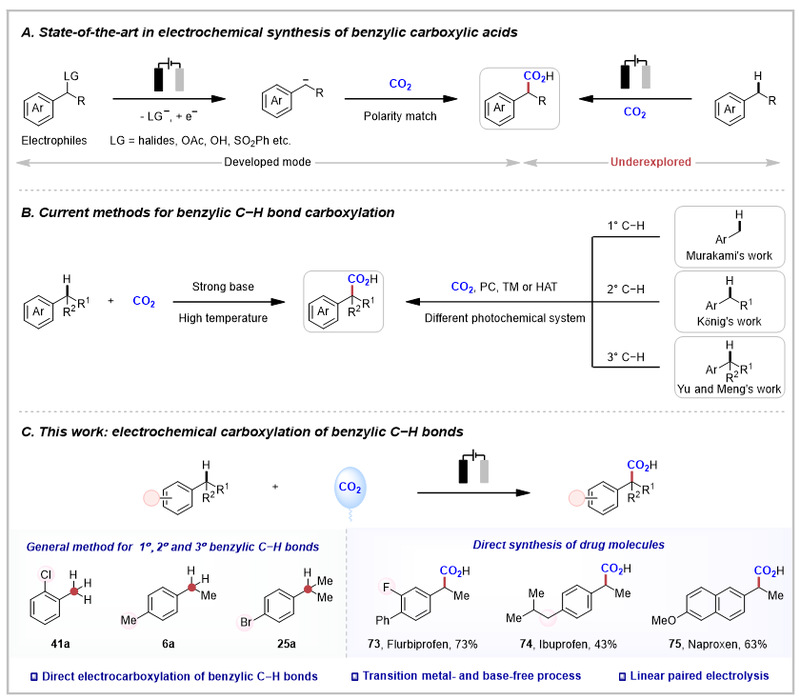
图1. 苄位羧酸的合成策略
南开大学化学学院元素有机化学国家重点实验室仇友爱课题组聚焦于有机电合成化学及资源小分子高效转化(Acc. Chem. Res. 2025, 58, 113-129; Nat. Catal. 2024, 7, 412-421; Angew. Chem. Int. Ed. 2025, 64, e202425634; Nat. Commun. 2025, 16, 2322; Angew. Chem. Int. Ed. 2022, 61, e202207746; Angew. Chem. Int. Ed. 2022, 61, e202210201; Angew. Chem. Int. Ed. 2023, 62, e202214710; Nat. Commun. 2022, 13, 3774; Angew. Chem. Int. Ed. 2022, 62, e202312803; Nat. Commun. 2024, 15, 2780; Nat. Commun. 2024, 15, 3832; Angew. Chem. Int. Ed. 2023, 62, e202306679; Angew. Chem. Int. Ed. 2023, 62, e202311941; Nat. Commun. 2023, 14, 6467; Angew. Chem. Int. Ed. 2022, 61, e202115178; Nat. Commun. 2024, 15, 5181; Chem. Soc. Rev. 2025, in press; CCS Chem. 2024, in press)。近日,仇友爱课题组报道了苄位C–H键电羧化反应(图1C)。该工作具有以下特点:a) 苄位C−H键的直接羧化反应,最大限度地提高了原子利用率;b) 提供了一种通用的方法,用于合成仲、叔及季碳苄位羧酸;c) 无需过渡金属和碱参与,具有良好的官能团耐受性;d) 直接合成药物分子,如非诺洛芬、酮洛芬和布洛芬等;e) 成功实现了复杂分子的后期修饰,表现出该线性成对电解反应的优异性。
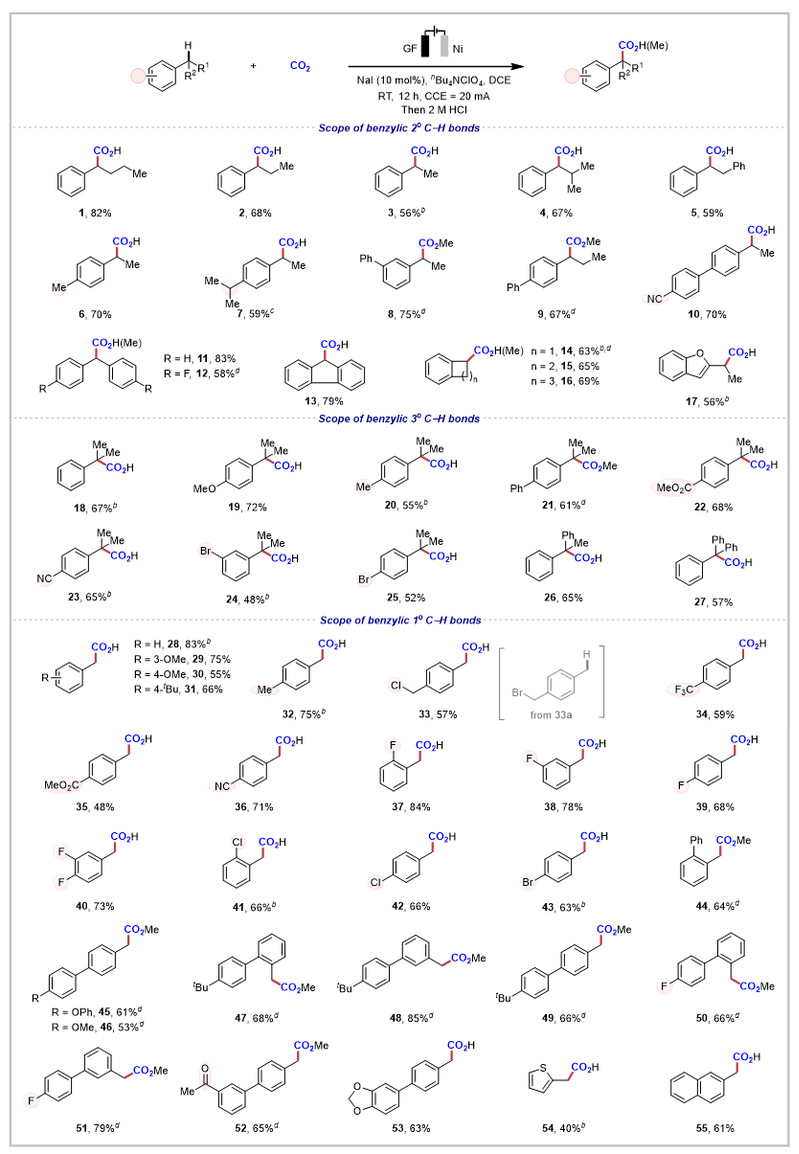
图2. 底物普适性研究
此方法适用于带有不同链长和不同空间位阻的底物(图2)。在具有多个苄基位点的底物,这种电化学策略也展示了高度的位点选择性(6,7和20)。一些电化学敏感的官能团,如酯基和氰基,得到了良好的耐受,为进一步应用提供了潜在机会。各类含有一级、二级和三级苄位C−H键的底物都能顺利得到苄位羧酸产物,并且杂环和稠合芳烃作为反应物时,能够与二氧化碳偶联,以中等产率得到54和55。
另外,该策略能够实现复杂分子的后期修饰(图3)。一系列具有生物活性的重要衍生物(如非诺贝特酸、奥沙普秦和胆固醇等)均可与二氧化碳发生羧化反应,成功制备出苄位羧酸类化合物56至68。含有酮基、噁唑、吡啶、酰胺、烯烃或乙酰基等敏感基团的底物均表现出良好的反应性,能以较高收率获得相应产物。
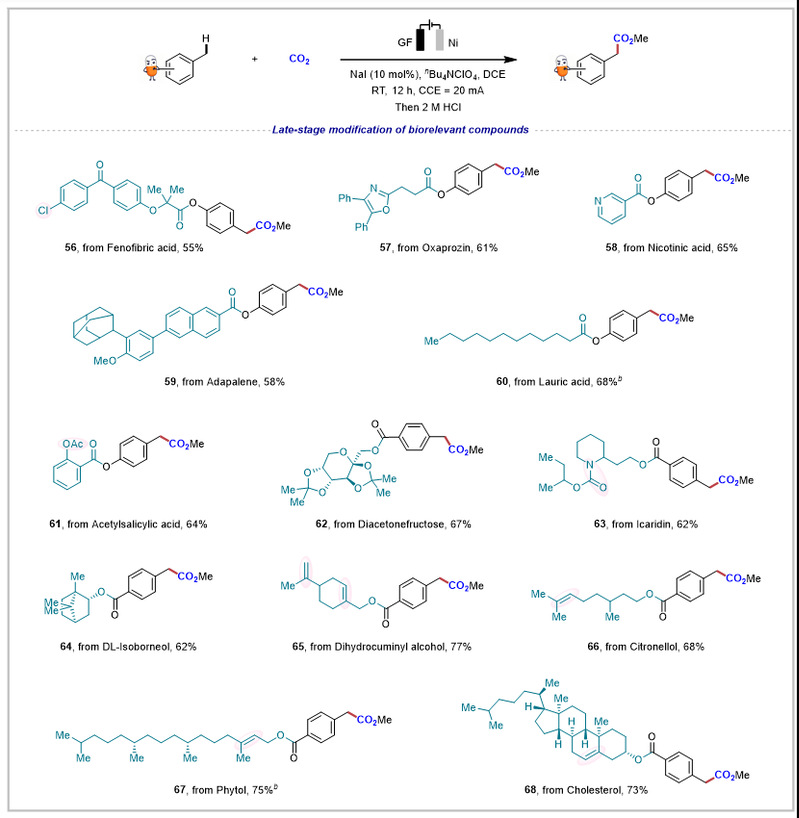
图3. 复杂分子后期修饰的普适性探索
该电化学方法被证实可直接高效合成多种常见药物分子,如非诺洛芬69、氟比洛芬73和萘普生75(图4)。需要特别指出的是,尽管1-乙基-4-异丁基苯存在两个苄位C(sp3)−H键,但空间位阻效应促使反应高选择性地生成布洛芬74。此外,瑞格列奈中间体76和西格列汀中间体77的成功制备,进一步印证了该电化学羧化策略的应用潜力。克级实验也证明了该反应具有优异的步骤经济性和原子经济性。
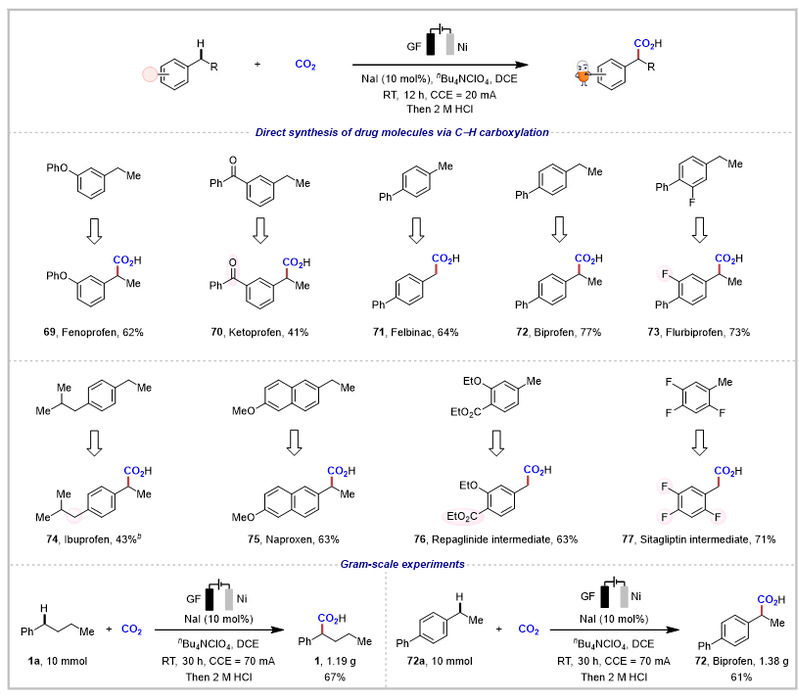
图4. 药物分子的制备及克级实验
作者对机理展开了细致的研究。首先,一系列的自由基捕获实验证明了该反应经历的自由基过程(图5A)。氘代产物的合成说明反应可能涉及碳负离子的生成(图5B)。控制实验表明在氩气氛围下可以得到氯化产物72a,且该产物在二氧化碳氛围下可进一步转化为羧化产物(图5C)。循环伏安实验表明,DCE可先发生还原反应,随后进一步被在阳极被氧化(图5D)。
接着作者提出了可能的反应机理:DCE获得一个电子形成氯阴离子,随后在阳极被氧化。氯自由基与底物I之间的发生的氢原子转移过程生成苄基碳自由基II和HCl。II通过氧化形成III,并与氯阴离子偶联生成苄基氯。原位生成的IV失去一个氯离子并还原生成V,CO2随后对其进行亲核攻击后获得最终产物。
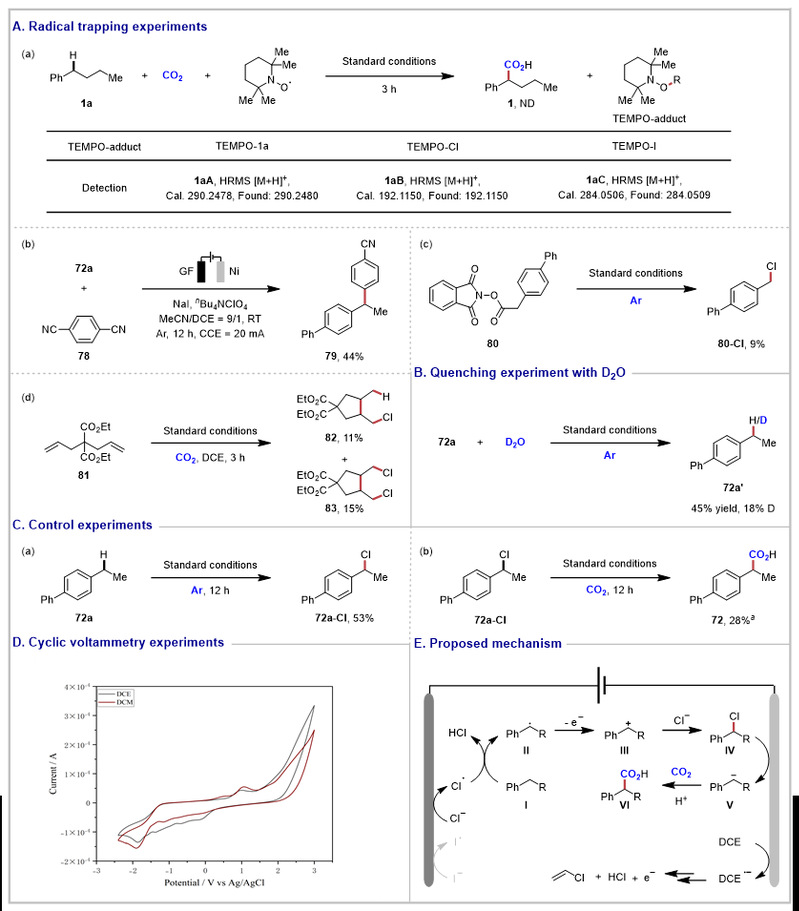
图5. 机理探究
小结
仇友爱团队发展了苄位C–H键电羧化反应,该方法在温和条件下进行,底物范围广泛。该方法为复杂分子的后期修饰以及苄位羧酸药物分子的直接合成提供了一条便捷且高效的途径。该工作得到了科技部重点研发专项、国家自然科学基金委、中央高校基本科研业务费及南开大学有机新物质创造前沿科学中心专项资金的支持。相关成果发表在Journal of the American Chemical Society 上。南开大学化学学院仇友爱研究员为通讯作者,博士生曾伟美为文章的第一作者。

关注“南开化学”微信公众号