
理解材料表面官能团的迁移行为是调控催化性能的关键。近日,南开大学孔祥蕾团队首次在水合氧化镧幻数团簇的结构和光谱研究中揭示了一种全新的羟基迁移机制——由摇摆振动诱导的羟基迁移。该机制能垒低(<12 kcal/mol),且无需断裂O-H键,为理解表面催化过程提供了全新的分子级视角。
羟基(OH)在众多多相催化反应中扮演着关键角色,其表面的迁移行为直接影响催化活性和选择性。传统的质子跳跃机制通常涉及质子的转移和O-H键的断裂。然而,对于表面OH基团是否存在其他更优的迁移路径,科学家们知之甚少。为了解决这一难题,南开大学研究团队巧妙地利用氧化石墨烯(GO)辅助激光烧蚀技术,成功制备并捕获了一系列结构稳定的 “幻数”水合氧化镧团簇( [(La₂O₃)n(H₂O)m(LaO)]⁺)。他们在实验室设计搭建的高分辨质谱(FT-ICR MS)- 红外光解离光谱(IRPD)系统上进行了一系列的实验,并结合理论计算,首次精确解析了这些团簇的稳定结构。
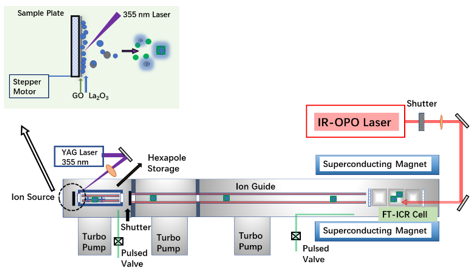
图1 实验使用的装置图

图2 实验中观察到的幻数团簇离子

图3 计算得到的幻数团簇离子(3,7)的多种构型
团队首先成功解析了(1,4)、(2,6)、(3,7)等多个幻数团簇的最稳定构型,发现所有团簇的光谱都在高波数区,从而确认其中的OH基团均以自由形式存在(未形成氢键),并可依据其配位环境分为三类。其中,对于(3,7)团簇,计算发现其最稳定结构 (3,7)-1 是一个低对称性的堆叠结构,并且存在一个与之能量完全相同的镜像异构体 (3,7)-1'。二者之间可能存在一个能量较低的、易于发生的相互转化路径。在意识到这一有趣的现象后,团队通过理论计算,深入揭示了这些团簇异构化过程的本质:一种由OH基团围绕金属中心的振动摇摆(Rocking Vibration) 所驱动的迁移。
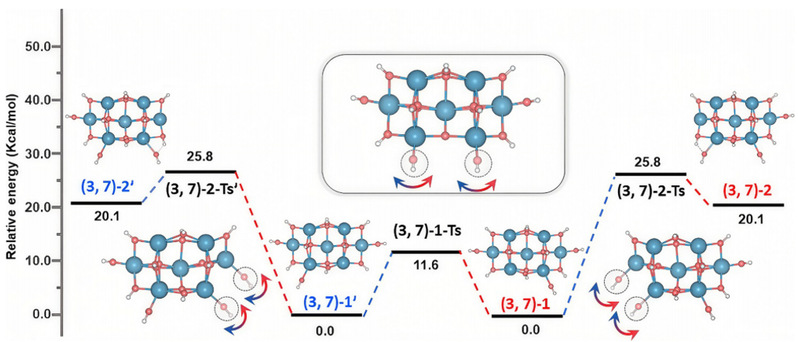
图4 摇摆振动诱导羟基的迁移过程与能垒(单位kcal/mol)
该过程最显著的特征是其极低的能量壁垒,经计算仅为约11.6 kcal/mol,低于离去-再结合的反应能垒。其中OH基团并非通过解离-再结合的方式,而是作为一个完整的单元,像传递接力棒一样,通过围绕金属-氧配位键的协同摇摆运动,从一个位点平滑地迁移到邻近位点。整个过程中,O-H键并未断裂,迁移涉及的是金属与氧原子之间离子-配位键环境的微妙调整。这与传统的“质子跳跃”机制存在根本性区别。后者本质是质子的迁移,必然涉及原有O-H键的断裂和新O-H键的形成,其反应坐标与O-H键的伸缩振动密切相关。而新机制的反应坐标则对应M-OH键的摇摆振动,整个过程是配位层结构的重组。此外,传统的质子跳跃通常需要一个预先形成的氢键网络作为“桥梁”来稳定过渡态;而振动摇摆机制在此体系中无需氢键的辅助即可高效发生。这些结果也暗示这一机制在各类金属氧化物表面可能具有更广泛的适用性和重要性。
这一成果近期发表在Journal of American Chemical Society上,文章的第一作者是南开大学化学学院已毕业的硕士研究生杨淑梅。本研究得到了国家自然科学基金、天津市自然科学基金、国家重点研发课题等多项基金的资助。
论文信息:
The Hydroxyl Migration Mechanism through Vibrational Rocking Unveiled from Magic-Numbered Hydrated Lanthanum Oxide
Shumei YangYameng HouShen BianKairui YangXianglei Kong

关注“南开化学”微信公众号