
来源:有机新物质创造前沿科学中心

1.研究背景
含有手性中心烷基链的氮杂芳烃广泛存在于药物、农药及天然产物中。该类结构的合成中,烷基链 α 位C-H键经去质子化后与亲电试剂发生不对称偶联,是最为直接的制备方法。然而,受限于 α 位质子酸性较弱的特性,现有方法通常仅适用于带有强吸电子基团的氮杂芳烃底物。对于不含吸电子基的未活化氮杂芳烃,去质子化需依赖强碱介导,这不仅易引发背景反应,还可能导致手性催化剂分解,为该类化合物的不对称催化转化研究带来巨大挑战。
对共轭烯炔进行氢金属化,可原位生成一类特殊的联烯丙基金属亲电试剂,进而在手性金属催化剂作用下,实现手性联烯结构的高对映选择性构建。然而,该策略目前仅适用于酸性较强的亲核前体;对于酸性较弱的未活化烷基氮杂芳烃,其去质子化需依赖强碱,而强碱生成的共轭酸酸性过弱,导致质子无法有效转移以产生关键亲电体,最终使该类底物难以兼容现有催化体系。
基于上述分析,通过特定方法增强烷基氮杂芳烃 α 位酸性,有望实现过渡金属催化下未活化烷基氮杂芳烃与共轭烯炔的不对称偶联反应,同时拓展两类反应体系的适用边界。
2.研究内容
南开大学化学学院、有机新物质创造前沿科学中心王晓晨教授课题组前期研究发现,硼催化剂可有效活化氮杂芳烃 α 位C-H键,增强质子酸性,而且,利用位阻调控可形成与底物的动态配位/解离平衡,以确保催化循环(J. Am. Chem. Soc. 2024, 146, 24663–24669)。以此为基础,王晓晨课题组在本文中报道一种硼/镍协同催化体系,首次实现了未活化2-烷基苯并噁唑与共轭烯炔的不对称偶联反应(图1)。
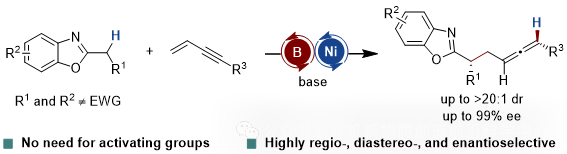
图1. 硼/镍协同催化非活化2-烷基苯并噁唑的不对称联烯丙基化反应
通过条件优化,确定以B[3,5-(CF3)2C6H3]3(LA1)作为路易斯酸催化剂,与由Ni(cod)2和手性Josiphos配体SL-J009-1原位生成的镍催化剂协同作用,实现高立体选择性转化。该反应展现出优异的底物普适性:无论是2-位带有不同烷基链或苯环含多种取代基的苯并噁唑,还是芳基、杂芳基及烷基取代的共轭烯炔,均能有效兼容,以较高收率和高立体选择性获得相应目标产物(图2)。
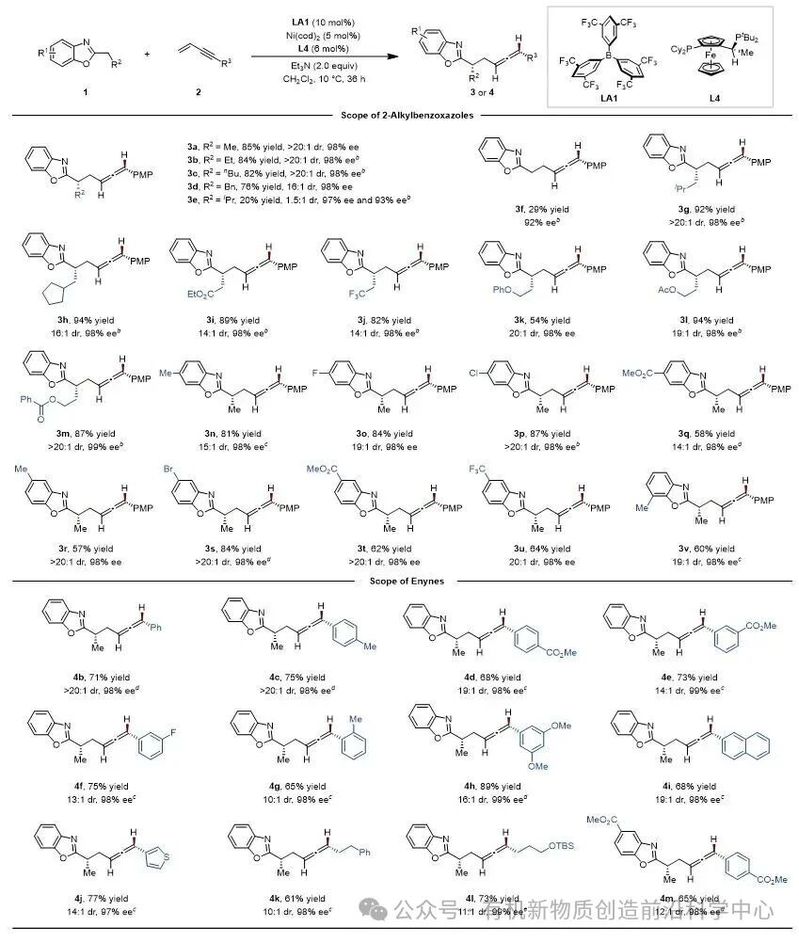
图2. 不对称联烯丙基化的底物范围
该联烯丙基化反应可顺利放大至克级规模,其轴手性中心能通过后续转化转变为中心手性,且反应并不会降低其非对映选择性与对映选择性(图3)。
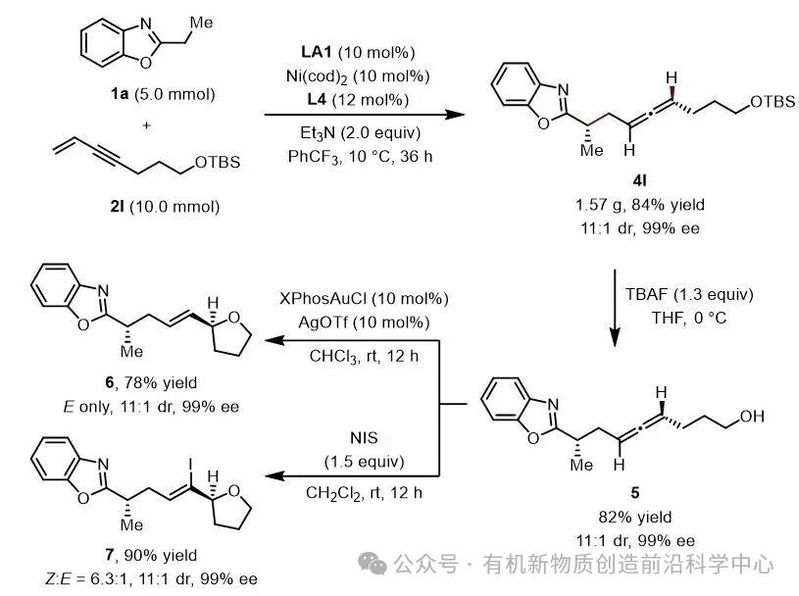
图3. 克级实验与衍生
基于相关文献报道,作者提出了可能的反应机理(图4):在硼催化循环中,硼催化剂通过配位增强苯并噁唑 α 位质子酸性,随后在碱的作用下发生去质子化,形成高活性亲核中间体。在镍催化循环中,零价镍首先与炔烃发生氧化加成,生成镍(II)-烯烃三元环中间体;该中间体被碱所形成的共轭酸质子化,进而生成关键的 π-联烯丙基镍亲电中间体。最终,亲核中间体与亲电中间体发生偶联反应得到目标产物,同时实现硼催化剂与镍催化剂的再生。
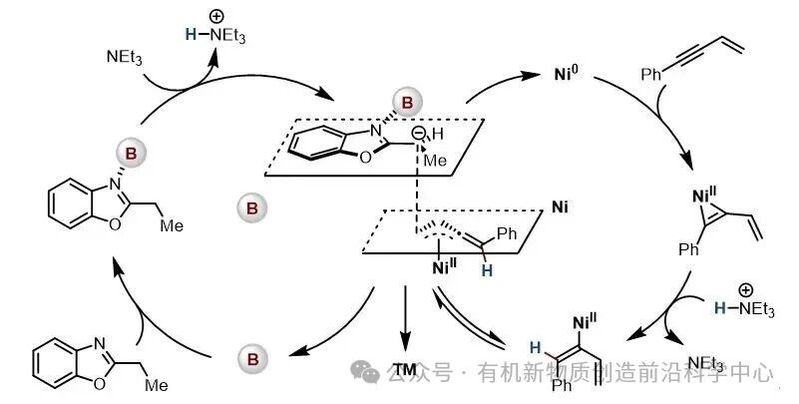
图4. 推测的反应机理
在此基础上,该课题组进一步将“硼–过渡金属”协同催化策略拓展至未活化氮杂芳烃的不对称烯丙基化反应。之前,该类反应中构建含两个相邻手性中心的产物报道很少,且局限于环状烯丙酯底物。研究发现通过合理调控硼催化剂结构,以B[3,4,6-F3C6H2]3(LA4)作为催化剂,并以[Pd(allyl)Cl]2与PHOX型配体原位生成的钯物种作为金属催化剂,成功实现了2-烷基苯并噁唑与链状1,3-二取代烯丙酯的不对称偶联反应。该催化体系底物适用范围广泛(图5)。
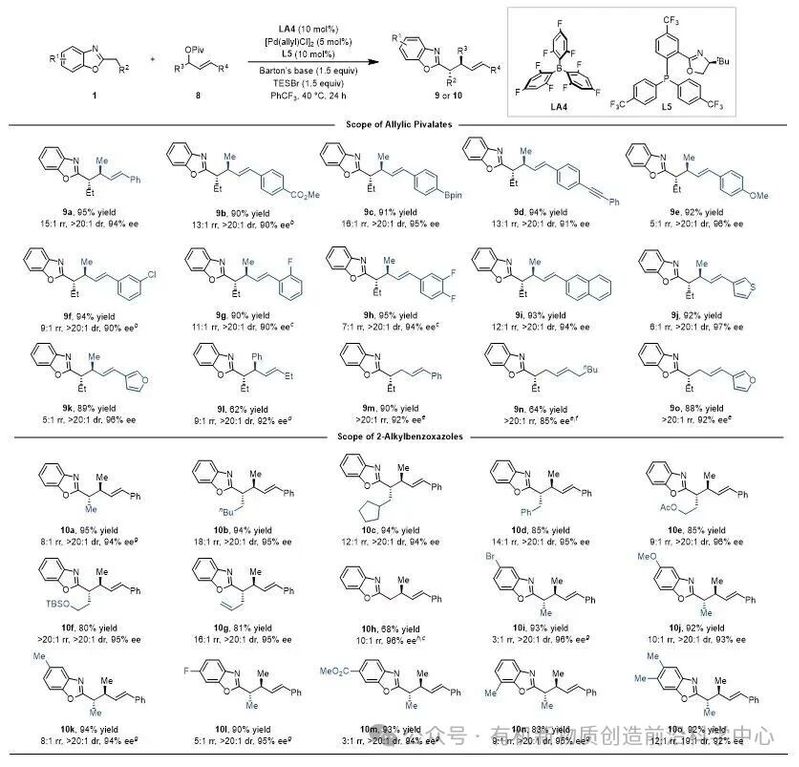
图5. 不对称烯丙基化反应的底物范围
综上所述,王晓晨教授课题组通过发展硼/过渡金属协同催化策略,成功实现了未活化2-烷基氮杂芳烃α位的不对称联烯丙基化和烯丙基化反应,为该类重要骨架的合成提供了高效、高选择性的新方法,同时也证明了硼/过渡金属协同催化策略在不对称合成中的广阔前景。
这一成果近期以“Borane/Transition Metal–Catalyzed Allenylic and Allylic Alkylation of Unactivated 2-Alkylbenzoxazoles”为题发表在《Journal of the American Chemical Society 》期刊上,王晓晨教授为论文通讯作者,博士研究生刘启飞为论文的第一作者,南开大学有机新物质创造前沿科学中心为论文通讯单位。该工作得到了国家自然科学基金和国家重点研发计划的经费支持。
3.文献信息
Borane/Transition Metal–Catalyzed Allenylic and Allylic Alkylation of Unactivated 2-Alkylbenzoxazoles, Qi-Fei Liu, Qian-Qian Peng, Sheng-Shi Hao, Lu Liu, Fu-Hao Zhang, Zhu-Jun Shi, Jian-Hua Xie, Chong-Ren Ai*, Xiao-Chen Wang*, J. Am. Chem. Soc. 2025, DOI: 10.1021/jacs.5c13835

关注“南开化学”微信公众号