
来源:ChemBeanGo
导语
传统光催化体系因无法协同调控激子与电荷载流子动力学而受到根本性制约,这种局限性阻碍了量子效率的提升和规模化应用。为解决此问题,南开大学汪清民课题组报道了一种基于聚庚嗪酰亚胺钾的光催化剂体系,该体系负载原子级分散的铱单原子位点,并修饰了末端氰基官能团,成功实现了激子介导和载流子介导路径的耦合。其中铱单原子位点有效缩小了单线态-三线态能隙,促进系间窜跃和三线态的生成,从而增强能量转移过程。同时,末端氰基构筑了电荷传输通道,加速了电荷分离和界面电子转移。而单原子与氰基的协同作用拓宽了光吸收范围。这种双路径设计增强了可见光下活性氧物种的生成,激子介导的能量转移主要生成单线态氧,而载流子介导的电子转移产生超氧自由基;这两个过程共同驱动实现可见光下的氧化偶联反应构建S-X键(X=P、C、S)。该催化体系在无牺牲剂或额外助催化剂的条件下实现,底物适用范围广,具备克级生产能力,并表现出5次循环的稳定性。该方法同时兼容单原子催化剂的克级制备,凸显了其实际应用潜力。相关研究成果发表于ACS Catalysis,DOI:10.1021/acscatal.6c00056。
研究背景与设计理念
全球化快速发展加剧了环境退化和资源短缺,对可持续技术提出了迫切需求。聚合物半导体因其可调的光电性能和高效的光生电荷生成能力,成为具有吸引力的光催化剂。其较低的介电常数强化了库仑引力,产生紧密结合的电子-空穴对,使单线态激子占主导地位。单重态可通过系间窜跃形成长寿命的三线态激子,参与能量转移过程。尽管激子和自由载流子均对催化活性有贡献,但多数策略仅侧重优化激子路径或载流子路径之一,忽略了它们之间的内在相互作用。这种孤立的方法忽视了潜在机制的协同本质,限制了性能提升。因此对激子动力学和电荷传输进行协同控制是突破效率瓶颈的核心挑战。
单原子金属位点的催化作用受金属中心和载体环境共同支配。单原子中心的价电子数会调控相关反应路径:缺价电子位点倾向于载流子介导的电荷转移,而富价电子位点倾向于激子主导的能量转移。若孤立看待这些调控因素,将会过度简化和模糊催化机制。为获得更全面的认知,必须同时考虑载体结构、电荷传输环境及单原子位点的原位演化过程。聚庚嗪酰亚胺钾具有合适的带隙和丰富的非配位氮基团,是锚定单原子的理想载体。然而,其在电荷-空穴分离方面的内在限制削弱了催化活性。引入末端氰基可通过内建电场强化载流子分离,建立高效的传输通道。而引入贵金属单原子位点可以引发重原子效应促进三重激发态的形成。基于这些认知,作者尝试通过双功能的设计耦合激子介导的能量转移与载流子介导的电子转移,从而实现同步优化。这种协同作用最终赋予体系卓越的光催化效率。
载流子与激子路径分别通过电子转移和能量转移将氧气转化为相应的活性氧物种(超氧自由基和单线态氧)。这两种氧物种均可作为好氧氧化偶联反应中的绿色氧化剂。因此,在单原子催化剂中协同调控激子与载流子路径,有望实现同时利用能量转移与单电子转移的可控光催化过程。
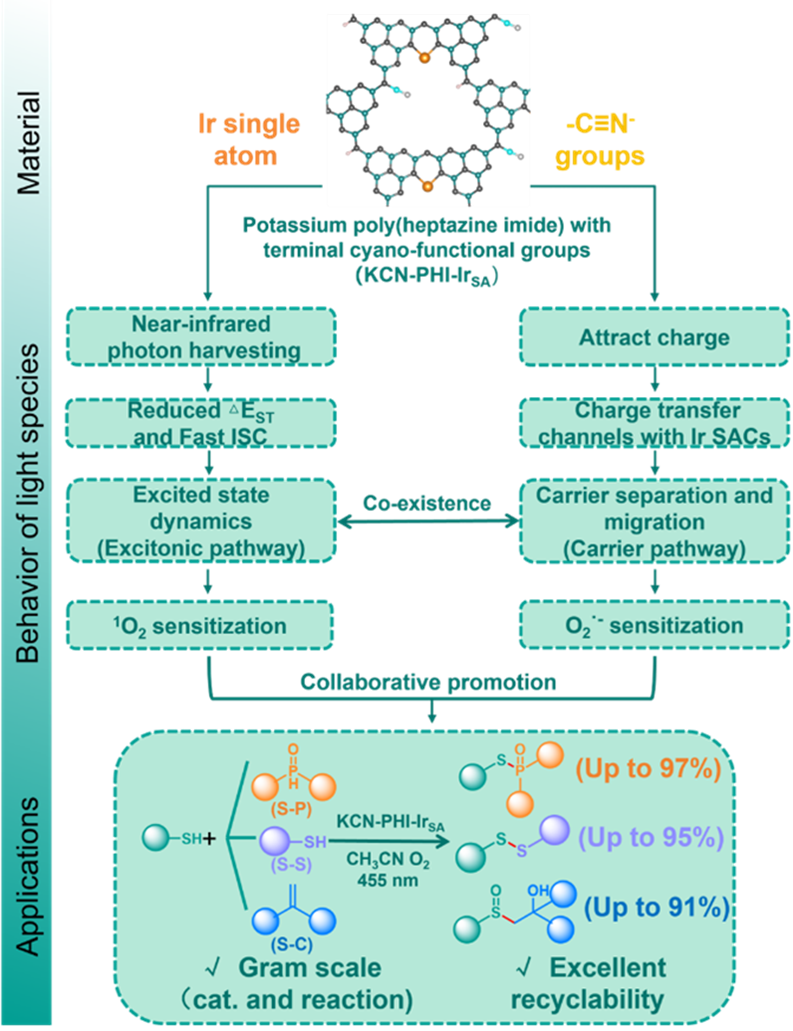
图1. 当前研究主题及其内在关联(来源:ACS Catal.)
基于以上,研究团队将铱单原子锚定在聚庚嗪酰亚胺钾上以强化激子效应,并引入末端氰基基团以促进骨架内的电荷传输(KCN-PHI-IrSA)。铱单原子中心作为重原子位点,降低了单重态-三重态能隙,增加三重态布居,强化激子介导的能量转移过程。同时,吸电子的末端氰基在七嗪环骨架中建立电荷传输通道,加速电荷分离和界面电子转移。该双通道的协同作用拓宽催化剂的光吸收范围,并进一步促进单线态氧和超氧自由基的生成,实现了高效、绿色的光催化氧化合成S-X(X = P, C, S)键。
前沿科研成果
催化剂的合成与表征
作者首先通过两步热缩合法合成具有末端氰基的聚庚嗪酰亚胺钾前体。具体为:通过氯化钾盐模板法将三聚氰胺热缩合,随后与硫氰酸钾共同煅烧引入末端氰基。得到前驱体后通过室温下的阳离子交换法引入铱单原子,制得KCN-PHI-IrSA。电感耦合等离子体质谱(ICP-MS)确认铱单原子的负载量为1.76 wt%。该合成方法支持克级(约1.49 g)规模制备。高分辨透射电子显微镜(HRTEM)显示KCN-PHI-IrSA由层状纳米晶体组成,且未观察到金属团簇。球差电镜图像(AC-HAADF-STEM)中呈现出代表铱单原子的一致且均匀分布的亮点,证明铱物种呈单原子分散状态。
通过X射线衍射(XRD)表征了KCN-PHI-IrSA的晶体结构。图谱显示,K-PHI和KCN-PHI在8.1°和28.4°处有明显的衍射峰,分别对应(100)晶面的面内有序和(002)晶面的层间堆叠。而KCN-PHI-IrSA的(002)峰相对于K-PHI和KCN-PHI移动至28.2°,表明层间距扩大,这归因于铱原子插层到庚嗪环之间。傅里叶变换红外光谱(FTIR)中,KCN-PHI和KCN-PHI-IrSA在2180 cm⁻¹处均出现归属于氰基基团的峰。¹³C固态魔角旋转核磁共振谱图中,114 ppm处的峰证实了末端氨基位点被氰基取代。
为了进一步验证KCN-PHI-IrSA的表面化学态与配位环境,作者进行了X射线光电子能谱(XPS)的表征。其中C 1s谱表明,引入铱单原子后,N=C-N(288.90 eV)和C=N(286.70 eV)峰发生正向偏移,表明铱位点的存在扰动了电荷分布。N 1s谱中,KCN-PHI-IrSA在399.70 eV处出现一个附加组分,归属于金属-氮(M-N)键。铱4f谱在61.36 eV和64.40 eV处出现双峰,表明Ir处于+3氧化态。随后铱L₃边的X射线吸收近边结构(XANES)和扩展X射线吸收精细结构(EXAFS)分析进一步确认了铱的电子态和配位环境。XANES光谱中,KCN-PHI-IrSA的白线强度介于IrCl₃和Ir箔之间。EXAFS傅里叶变换光谱在1.64 Å处显示主峰,归属于Ir-N配位,不存在Ir-Ir配位,表明Ir原子呈孤立状态。拟合结果揭示Ir-N配位数为2.1,表明Ir单原子与两个氮原子配位。小波变换(WT)分析进一步证实KCN-PHI-IrSA中缺乏Ir-Ir键。
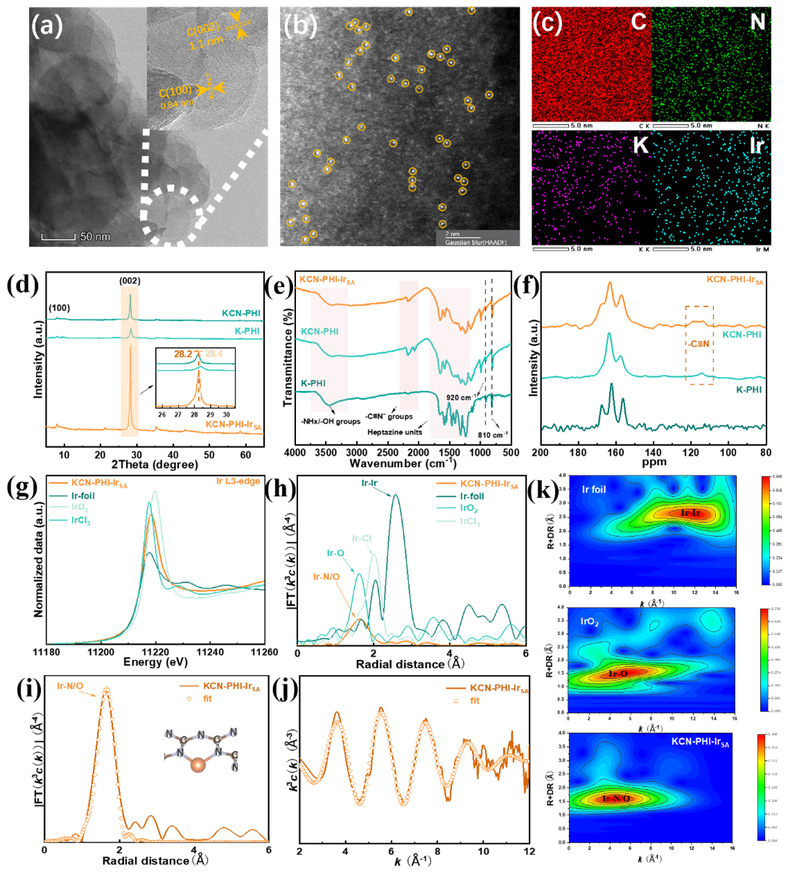
图2. KCN-PHI-IrSA的形貌表征(来源:ACS Catal.)
单原子与末端氰基共同调控能带结构
紫外-可见漫反射吸收光谱(DRS)表明,K-PHI的吸收边缘在445 nm。末端氰基的引入使KCN-PHI的吸收边缘红移至486 nm。进一步引入Ir单原子后,KCN-PHI-IrSA在约1.49 eV处出现带内态(IBS*),促进了n→π*跃迁。Tauc图计算得出K-PHI、KCN-PHI和KCN-PHI-IrSA的带隙能量,并通过价带XPS确定其价带的位置。因此得出引入的氰基和单原子的相互作用调节了能带结构,增强的导带电子还原能力使其能够将氧气还原为超氧自由基(-0.35 V)。密度泛函理论(DFT)计算指出,理想K-PHI的带隙约为2.17 eV。末端氰基取代加强了庚嗪单元间的π共轭,带隙缩小至约1.77 eV 。Ir原子与七嗪环中的N配位,Ir 5d轨道与N 2p轨道杂化,产生浅层带隙内态,有效带隙降至约0.54 eV,对应于紫外光谱中观察到的IBS*特征。
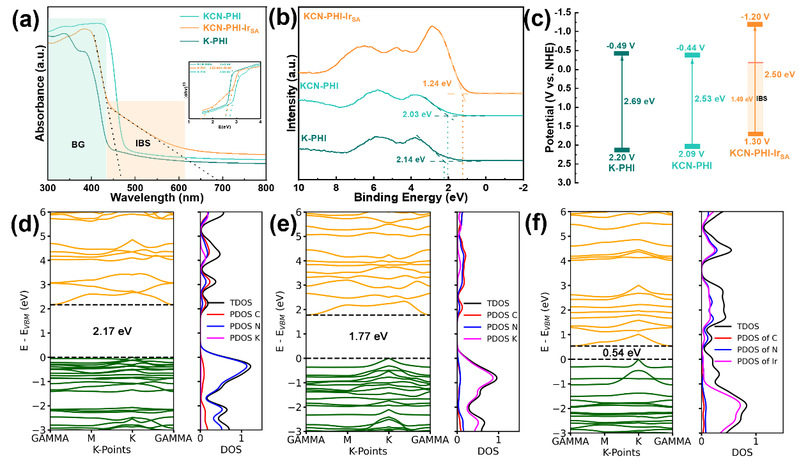
图3. 催化剂的能带结构(来源:ACS Catal.)
激子与载流子传输动力学
激子与载流子传输动力学是评估光催化机制的关键参数。作者通过稳态和时间分辨磷光光谱分析了光激发体系中的激子过程。对荧光和磷光的峰进行归一化以评估单重态-三重态能隙(ΔEST)。KCN-PHI-IrSA的ΔEST(0.314 eV)显著低于KCN-PHI(0.467 eV)和K-PHI(0.350 eV),证实单原子铱增强了系间窜跃过程。而氮气氛围下磷光强度显著增强,确认该体系高效布居了三线激发态。时间分辨磷光衰减曲线显示KCN-PHI-IrSA的平均磷光寿命最长(17.91 μs)。通过变温稳态荧光光谱(PL)和拟合分析,证明KCN-PHI-IrSA的激子结合能(Eb)为97.25 meV,高于KCN-PHI(57.46 meV)和K-PHI(62.03 meV),证明Ir单原子通过增强库仑相互作用强化了激子限域。
随后作者进行改性后催化剂的载流子动力学探究。常温下的稳态荧光光谱显示KCN-PHI-IrSA的荧光强度显著猝灭,瞬态荧光寿命缩短至0.42 ns,证明电荷载流子分离得到改善。光照下的电子顺磁共振(EPR)光谱中,KCN-PHI-IrSA在g = 2.0042处表现出最强的洛伦兹信号,证实了其优越的光生电荷载流子生成效率。开尔文探针力显微镜(KPFM)显示,在暗态下KCN-PHI-IrSA的表面电势差比KCN-PHI和K-PHI更负,表明形成了更强的内建电场。光照下,KCN-PHI-IrSA表现出更负的表面光电压(SPV)变化,表明大量光生电子向催化剂表面迁移和积聚,促进表面的单电子转移过程。
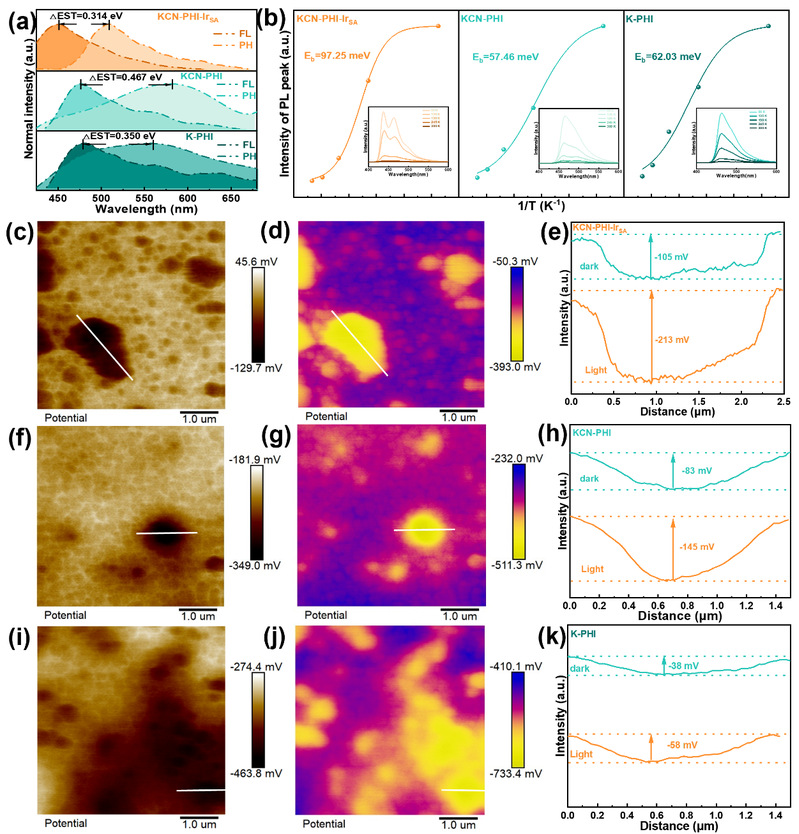
图4. 激子与载流子的光化学表征(来源:ACS Catal.)
活性氧的双通道生成机制
为了评估铱单原子与氰基功能化体系对活性氧生成路径的协同增强作用,作者使用5,5-二甲基-1-吡咯啉N-氧化物(DMPO)和2,2,6,6-四甲基哌啶(TEMP)分别作为超氧自由基和单线态氧的探针进行EPR自旋捕获实验。光照下,KCN-PHI-IrSA产生最强的TEMP-¹O₂加合物信号和DMPO-O₂·⁻信号。对苯醌(p-BQ)猝灭实验证实¹O₂完全由激子介导的能量转移产生,而O₂·⁻由载流子主导的电子转移产生。这些结果表明铱原子和末端氰基在增强激子效应和电荷分离效率方面具有关键作用。随后的准原位XPS揭示,光照时KCN-PHI-IrSA的C=N峰(285.99 eV)向高结合能方向偏移,表明光生空穴向末端氰基转移。同时,铱4f谱向低结合能方向偏移,表明电子在铱单原子位点富集,导致其氧化态低于+3,从而促进电子从铱转移至氧气。DFT电荷密度差图谱支持了这一结论,显示激发态下电子向铱配位环境聚集,有利于O₂吸附和还原。
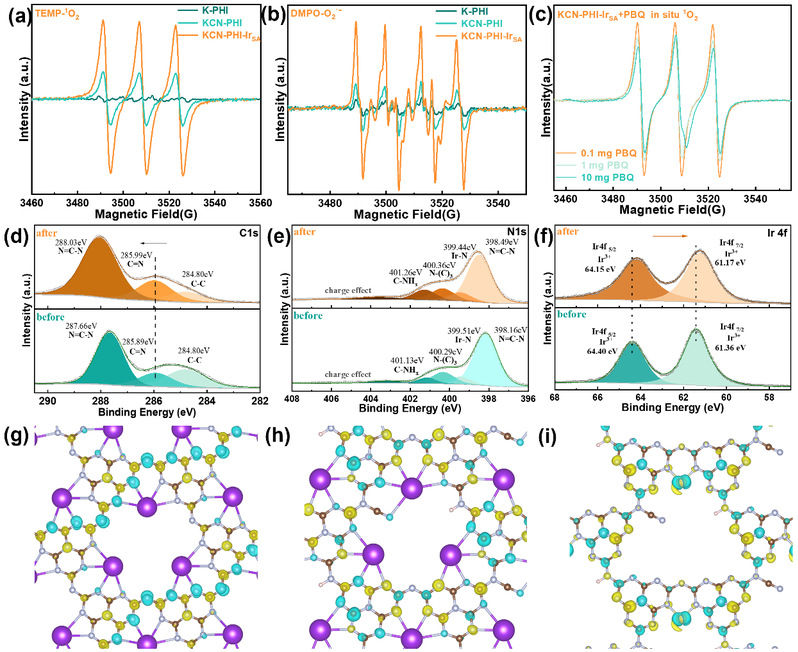
图5. KCN-PHI-IrSA体系中活性氧的生成机制(来源:ACS Catal.)
KCN-PHI-IrSA光催化氧化合成S-X键的性能研究
作者以4-甲氧基苯硫酚和二苯基氧化膦为模版底物,在450 nm光照下反应8小时,目标产物硫代次膦酸(S-P键)的转化率达到97%。条件优化中发现Ir单原子负载量过高(4.4 wt%)或过低(0.39 wt%)均会导致产率下降。在200 mg KCN-PHI-IrSA的克级实验中,获得了84%的分离产率,计算出的TON和TOF分别为330.3和27.5 h⁻¹。反应对带有多种供电子基团和吸电子基团的芳基硫醇表现出良好的耐受性。萘硫醇、杂环硫醇(吡啶、噻吩)以及苯硒酚均能有效参与偶联反应。二苯基氧化膦衍生物同样具有极佳的官能团兼容性。经过5次催化循环,催化剂活性(产率81%)和结构特征保持完好。
随后使用KCN-PHI-IrSA在氧气饱和的乙腈中,催化4-甲氧基苯硫酚与1,1-二苯基乙烯合成β-羟基亚砜(S-C键)。在450 nm的光照下5小时转化率达到92%。克级实验分离产率为73%。该反应体系对含多种取代基的硫酚和1,1-二苯基乙烯衍生物均表现出中等到极高的产率(43%–92%)。5次循环后仍保留82%的产率。
通过KCN-PHI-IrSA光催化4-甲氧基苯硫酚发生氧化偶联生成二硫化物。450 nm光照8小时后转化率达到95%,同时以86%的产率实现克级制备。底物拓展表明带有供电子基团的芳基硫醇获得71%–95%的产率。杂环硫醇以及异硫源二硫化物的合成也取得良好的结果。
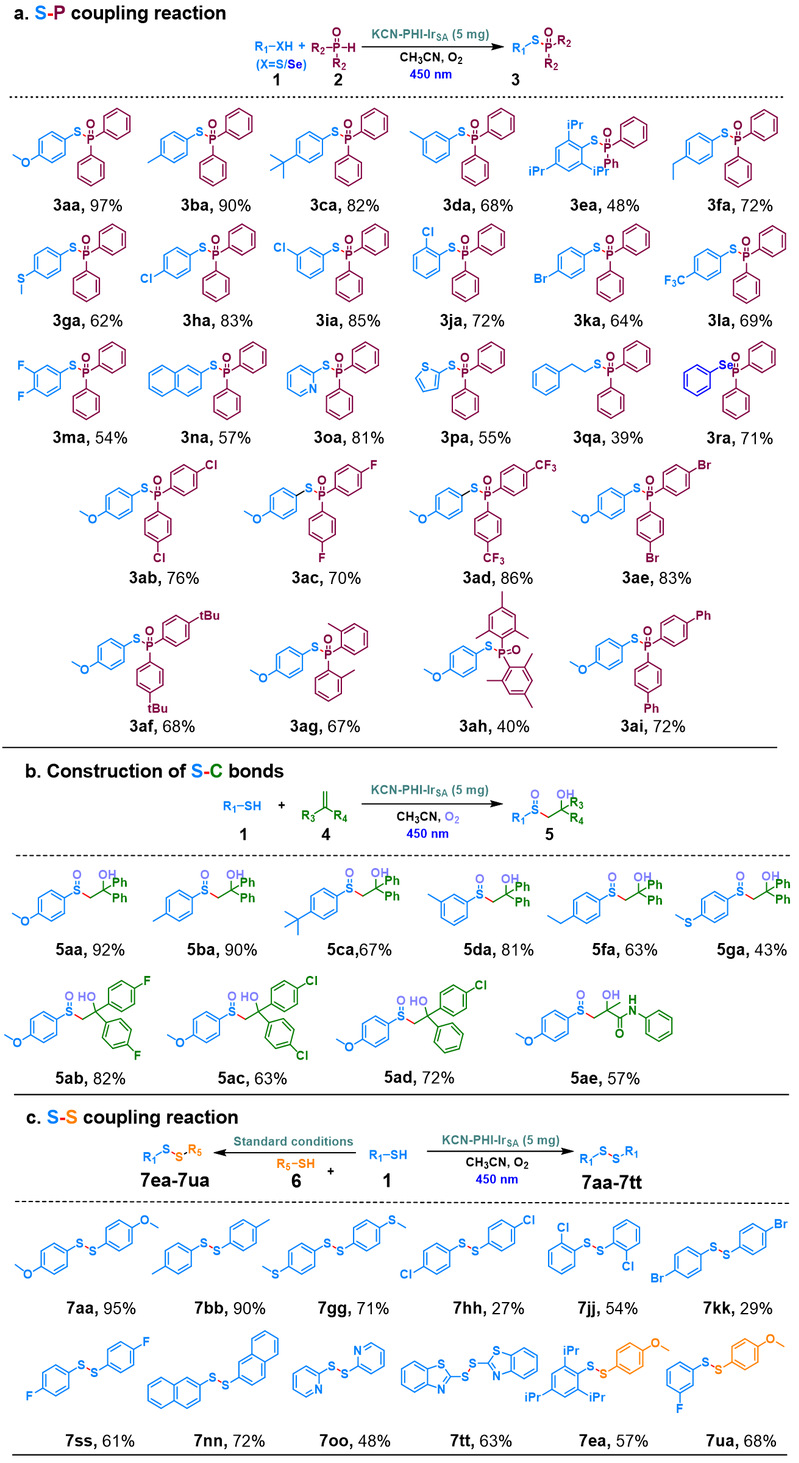
图6. KCN-PHI-IrSA光催化合成S-P键、S-C键和S-S键(来源:ACS Catal.)
KCN-PHI-IrSA光催化氧化反应机理研究
控制实验表明,加入自由基抑制剂TEMPO导致产率显著下降,证实了自由基介导的反应路径。加入超氧自由基猝灭剂(p-BQ)和单线态氧清除剂(NaN₃)均导致产率大幅下降,证明¹O₂和O₂·⁻的协同作用是驱动反应的关键。加入底物和光照的EPR实验显示,原有的g = 2.0042信号消失,同时出现g = 2.0031的新信号,证明光激发的KCN-PHI-IrSA*被硫酚底物猝灭,并形成KCN-PHI-IrSA·⁻自由基物种。在包含底物的原位EPR自旋捕获实验中,单线态氧和超氧自由基的信号均未检测到,表明S-X键的形成需要同时消耗这两种ROS。热力学计算结果显示,硫酚(-SH)与¹O₂反应的吉布斯自由能变化为-52 kJ/mol,明显优于与O₂·⁻反应(+20 kJ/mol),说明-SH优先与¹O₂反应。相反,硫醇阳离子(-SH·⁺)与O₂·⁻反应的吉布斯自由能为-669 kJ/mol,远低于与¹O₂反应(+319 kJ/mol),表明-SH·⁺优先与O₂·⁻反应。
因此作者提出双通道反应机制:可见光照射下,KCN-PHI-IrSA被激发。在电子转移途径中,定位在末端氰基上的光生空穴氧化硫醇底物生成硫醇阳离子(-SH·⁺)。光激发的催化剂被还原猝灭形成KCN-PHI-IrSA·⁻,随后在Ir单原子位点将O₂还原为O₂·⁻。O₂·⁻进而将-S·⁺还原为硫自由基(-S·)。同时,在能量转移途径中,强激子特征促使催化剂经历高效的系间窜跃过程并形成三线态,将能量转移至基态氧气生成¹O₂。¹O₂激活-SH形成-S·。产生的-S·自由基与反应底物交叉偶联,或二聚成中间体,最终生成含S-X键的产物。
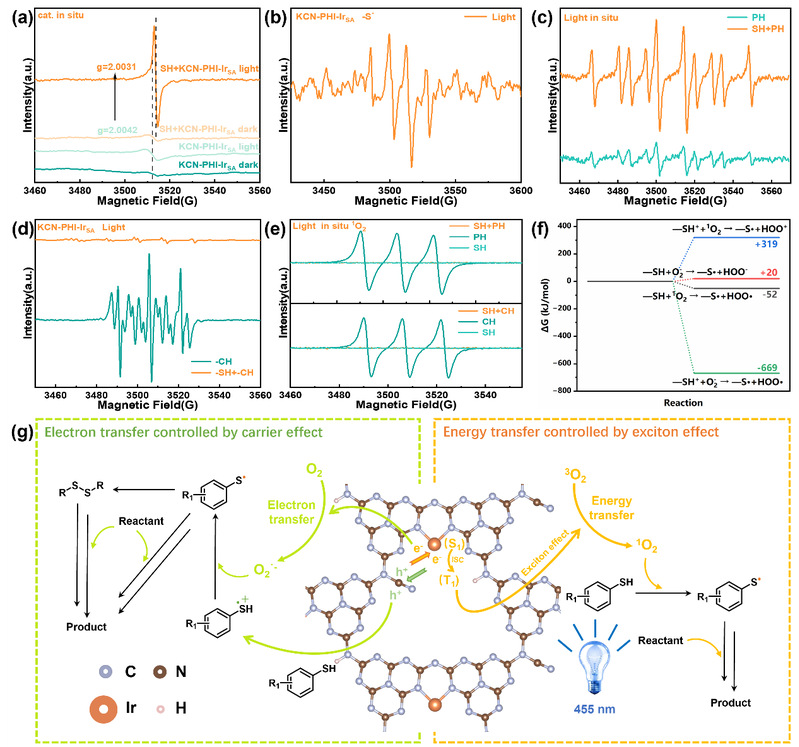
图7. KCN-PHI-IrSA光催化氧化的机制研究(来源:ACS Catal.)
本文提出了一种新颖的双功能策略,将铱单原子与末端氰基整合到K-PHI框架中,协同优化激子效应与载流子动力学。通过系统表征与理论分析,发现铱原子通过促进系间窜越来增强激子过程,而氰基则通过建立高效电荷转移通道促进电子分离与传输,并与单原子位点之间生成内建电场——导致电子迁移至铱位点,空穴则富集于末端氰基。两者之间的相互作用能够有效提升光吸收能力。这种双重调控实现了高效的双路径活性氧生成,在无需助催化剂或牺牲剂的绿色条件下,实现需氧氧化偶联构建S-X(X=P、C、S)键。原位电子顺磁共振与密度泛函理论计算共同阐明了S-X键的形成机制,证实单线态氧与超氧自由基均被消耗以生成关键的硫醇自由基中间体,进而驱动反应进行。该催化剂的可规模化合成凸显了其工业应用潜力。本研究为同步优化激子与载流子效应、协同利用双路径机制实现有机转化提供了创新策略。
鸣谢:本篇工作通讯作者为汪清民教授。南开大学博士研究生袁菲为该论文的第一作者。清华大学博士研究生孟祥泽为第二作者,完成本研究的计算部分工作。感谢刘玉秀和宋红健老师在实验中的指导和帮助。上述研究工作得到国家重点研发计划项目(2023YFD1700700)的资助。

关注“南开化学”微信公众号