
来源:BioPeers
蛋白质如何从一条无序的氨基酸链,折叠成精确的三维结构,这一直是结构生物学的核心谜题。在这个过程中,会形成一些稍纵即逝的“中间状态”,它们如同戏剧的幕后彩排,虽不登场,却决定了演出的成败。捕捉这些中间态对理解蛋白质功能及相关疾病至关重要,但实验手段难以捕获,而分子动力学模拟虽能生成海量原子运动数据,传统分析方法却往往“先入为主”——先基于几何结构猜测状态,再分析其动力学行为。这种“状态中心”的范式可能导致动力学信息的丢失,甚至掩盖了真实的折叠路径。
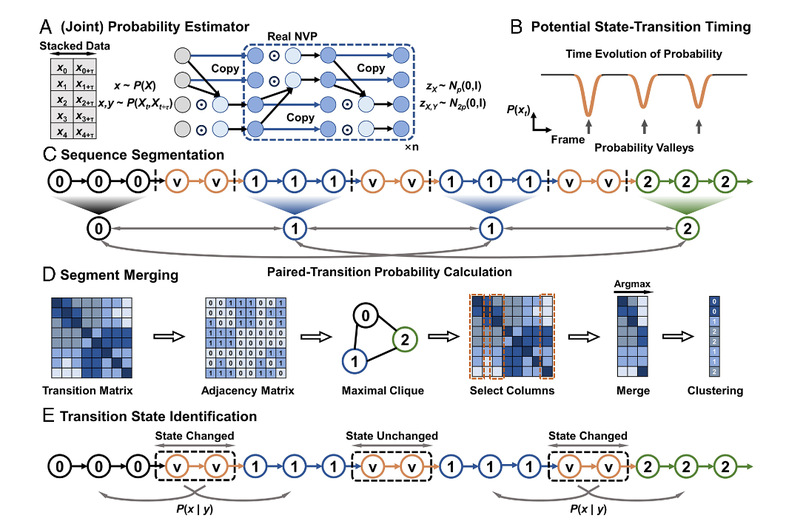
2026年3月6日,南开大学化学学院的邵学广教授/付浩浩副研究员/蔡文生教授等在《美国国家科学院院刊》(PNAS)上发表了题为《基于AI的条件转移聚类揭示蛋白质折叠中隐藏的中间状态》的研究论文。该团队提出了一种名为“条件转移聚类”的全新AI框架,开创性地将分析视角从“状态中心”转向“动力学中心”。这意味着,他们不再预先定义状态,而是先让AI学习系统内在的动力学规律,再根据“动力学隔离”的原则,让蛋白质的“状态”本身从数据中自然浮现。

为了验证CTC的效果,研究人员首先在具有双势阱(图A)和四势阱(图F)的简化模型上进行测试。对于双势阱系统,CTC通过分析轨迹随时间变化的概率分布,精准地识别出每次跨越能垒的“稀有事件”,并将轨迹划分为不同片段(图B)。通过计算片段间的转移概率,系统自然地聚合成了两个符合物理直觉的状态(图D-E)。在四势阱系统中,通过调整观测时间窗口(滞后时间),CTC成功捕捉到了动力学层次:短窗口下,四个状态清晰可辨;随着窗口拉长,快速平衡的相邻状态逐渐合并,最终整个系统被视为一个整体(图G-M),完美复现了自由能 landscape 的层级结构。
随后,CTC被应用于DE Shaw研究所提供的12条蛋白质折叠模拟轨迹。结果显示,在传统认为“双态”折叠的WW结构域中,CTC成功解析出了两个介于折叠与去折叠之间的中间态(a1, a2)。进一步分析发现,这些中间态虽然整体结构接近天然态,但在局部环区的二面角以及非天然接触上存在特征性差异(图A)。对于α/β蛋白NTL9,CTC识别出的四个状态(图B)中,两个中间态(b1, b3)分别表现出C端或N端螺旋的部分形成,这与文献中报道的中间体特征高度吻合。而在更复杂的蛋白G变体中,CTC甚至区分出了13个不同的构象状态(图C),其中包含多个与理论计算预测完全吻合的、具有特定缺陷的“近天然态”,如螺旋不完整或β-片层错位。
为了量化CTC的优势,研究者将其与传统马尔可夫模型及深度学习VAMPnets等方法进行了对比。在2纳秒的短滞后时间下,CTC在所有测试体系上都获得了最高的“亚稳性”分数,证明其能更有效地识别短暂的中间态,减少边界“往返”噪音的干扰。进一步的Chapman-Kolmogorov检验也证实,CTC构建的模型在2纳秒的短时间尺度上就具备良好的马尔可夫性,动力学自洽性优异。
CTC不仅能“定格”状态,还能“抓拍”过渡态。在双势阱模型中,CTC准确定位到了势能面上的鞍点(图A)。在蛋白质体系中,通过分析状态变化前后的“概率谷”区域,CTC识别出的过渡态结构(图E-G)恰好位于自由能垒的“山脊”之上(图B-D)。这些结构揭示了折叠过程中关键的成核事件,例如WW结构域中N端或C端β-片层的优先形成,为绘制完整的折叠路径提供了关键坐标。
基于识别的状态和过渡态,CTC最终绘制出定量的折叠“交通网络图”。对于WW结构域,一条从去折叠态直达天然态的主干道(ΔG‡=19.35 kJ/mol)主导了折叠过程(图A)。而NTL9则存在竞争路径,通过中间态的“换乘”比直达路线更为高效(图B)。对于蛋白G,CTC构建了包含多条路径的网络(图C),并计算发现,从去折叠态出发,经由c4、c5、c9再到天然态的路径是一系列能量障碍最低的“最优选择”,为理解复杂蛋白质的折叠机制提供了前所未有的动态蓝图。

关注“南开化学”微信公众号