
来源:有机新物质创造前沿科学中心

01 研究背景
受绿色植物光合作用的启发,利用太阳能驱动CO2转化为高附加值化学品,是实现能源可持续与环境协调发展的关键路径 。然而,由于CO2分子热力学稳定性和高键解离能使得高效光催化活化变得困难。常规的光催化剂通常仅能吸收紫外和可见光,从而导致在太阳光谱中占比近半的红外波段能量未被充分利用。为了解决这些局限性,光热催化通过利用局域表面等离子体共振(LSPR)、非辐射复合和振动弛豫将光子能量转换为声子能量,已成为一种有效策略。如示意图1所示,光热催化因其独特的能量转换途径而区别于传统热催化和光催化。具体而言,“光子→声子”过程通过半导体中的非辐射弛豫或共轭体系中的分子振动加热,促进光热转换。这种协同机制结合了“光子+声子”效应,通过降低活化能和减少总体能耗,使催化反应能够在温和条件下进行。因此,开发同时具备高效光捕获和快速光热转换能力的光热催化系统,仍具有重大挑战。
理想的光热催化剂不仅需要光敏剂来捕获光,需要催化中心来活化反应,而且关键的是需要它们之间高效的电子通路。当前体系常常存在空间分离问题,导致电荷或能量转移效率低下、光热效率降低以及催化活性欠佳。金属有机框架(MOF)为解决上述问题提供了极具前景的平台。其周期性和可调结构使得能够:(i) 在分子尺度上对多种功能基团进行精确构筑;(ii) 抑制均相体系中常见的聚集和失活;以及 (iii) 增强催化剂的可回收性和循环利用。此外,部分具有半导体性质的MOF还能通过非辐射衰减途径,伴随晶格声子释放来产生强烈的局域热能,从而极大地促进光热协同催化效率。[Ru(bpy)3]2+配合物 (bpy=2,2′-联吡啶) 是一种经典的光敏剂,具有强的可见光吸收和长激发态寿命。然而,其在框架材料中的应用主要聚焦于光催化中的电子跃迁,往往忽视了其通过声子介导弛豫作为局域热源的潜力,这大大限制了其能量利用效率。
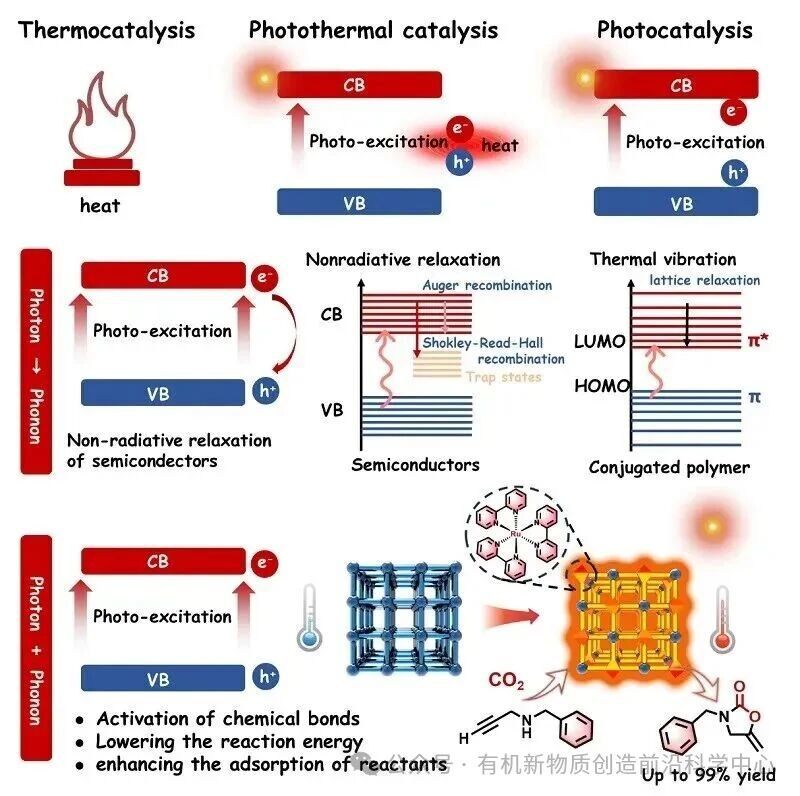
示意图1. MOF材料光热催化的示意图
02 研究内容
近日,南开大学化学学院、有机新物质创造前沿科学中心赵斌教授、阳科研究员联合深圳大学陈智副研究员报道了一种三维MOF,即{[Ru(BPDC)3Zn2(H2O)3]·4H2O}n(GIA-RuZn),通过简便的一锅原位策略合成。与需要多步配体预合成的传统方法不同,该方法将[Ru(bpy)3]2+光热单元和Zn(II)催化位点直接嵌入到单一集成框架中。这确保了热源与活性位点之间达到原子尺度的接近,从而最大限度地减少了热耗散。所得结构相互穿插的二维层进一步编织成三维架构,在水中对强酸和强碱条件(1M HCl和2M NaOH)表现出优异的化学稳定性。此外,GIA-RuZn表现出宽光谱光吸收和超快光热转换性能,在808 nm激光照射(0.8 W cm-2)下,1.3秒内达到155.7 °C。在20 W白光照射下,能够高效催化炔丙胺与CO2的羧化环化反应,生成2-噁唑烷酮类化合物,产率高达99%,TON达到556。机理研究表明,GIA-RuZn同时活化了两种底物并降低了环化步骤的能垒。
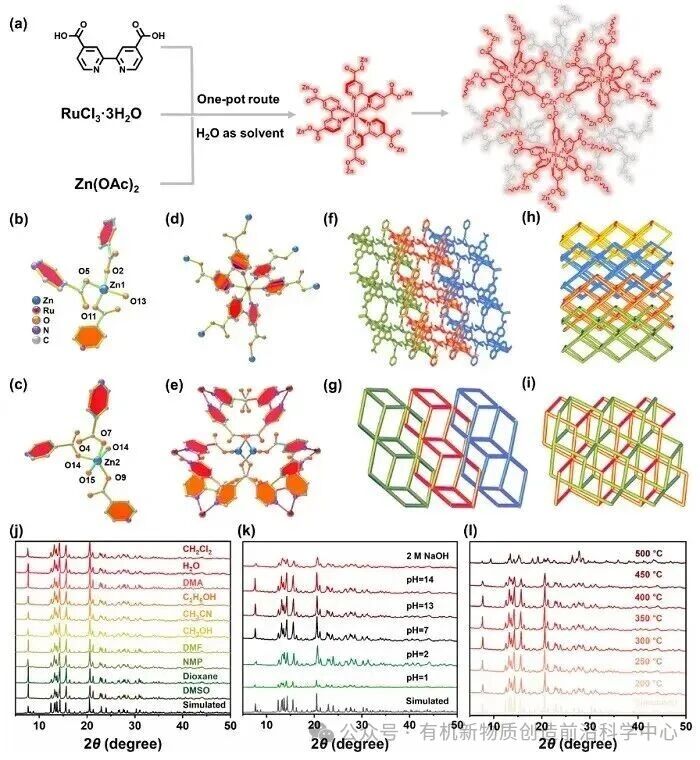
图1. GIA-RuZn催化剂制备流程及结构图和稳定性表征
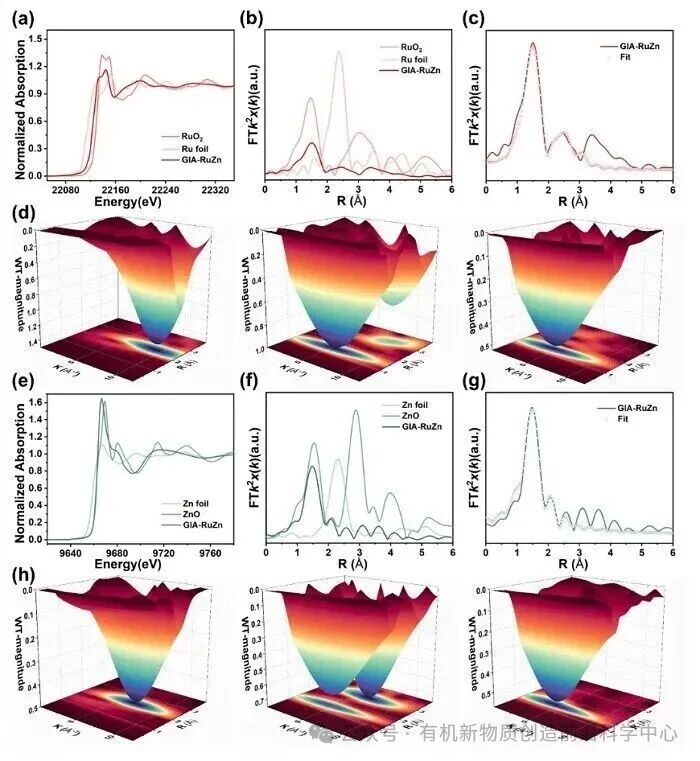
图2. XAFS对GIA-RuZn配位环境及金属价态的分析
如图1a所示,将RuCl3·6H2O,Zn(OAc)2,和H2BPDC配体直接溶于10 mL水中,在一锅法溶热反应成功合成了GIA-RuZn。单晶 X 射线衍射(SCXRD)表征揭示,该化合物属于单斜晶系,结晶于手性空间群C2。不对称单元分析表明,该材料由1个Ru2+中心、2个具截然不同配位模式的Zn2+离子组成。相邻的金属中心依靠具有长程双功能的BPDC2-配体相互交替桥联,沿(110)晶面无限横向延伸,形成了具典型1D微孔通道特征的2D层状网络结构(图1f)。游离的客体水分子则规整排列于中部通道中。拓扑结构学简化分析表明,若将Ru2+、Zn2+节点及三分叉的 BPDC2-桥联配体均简化为三连接拓扑节点,该2D层状网络呈现出 {6.102} {6.8.10} {6.82}的Schläfli拓扑。其2D层的底部通道与相邻2D层的顶部通道互穿。从横截面微观视角来看,2D层的底面层(4.82拓扑类型)与邻近2D层的顶面晶格相互交叉,自发编织成一个坚固的三维编织网络。这种独特的二维互锁结构以平行互锁的方式层层堆叠,最终在宏观上构筑成具有独特二维到三维(2D-to-3D)交织穿透特征的3D刚性多孔晶格(图1f-1h)。将该MOF分别浸泡于不同的常规有机溶剂和PH溶液中进行溶剂稳定性考察。PXRD 结果表明,该材料在多种溶剂和pH值1 至 14甚至2M NaOH强碱溶液中中皆保持结构完好,在高达450 °C的高温下,该MOF框架结构依然完好,未发生坍塌或碳化。
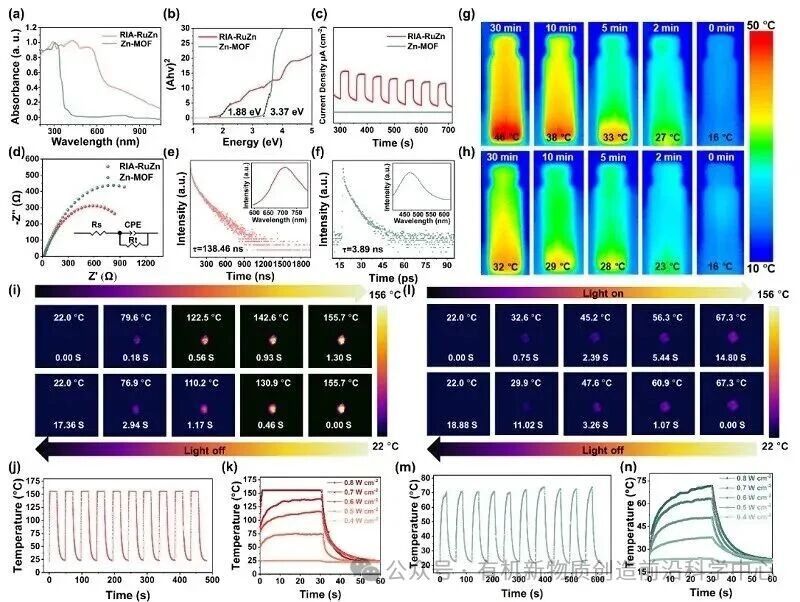
图3. GIA-RuZn和Zn-MOF光电性能与光热性能表征
对材料的光学、光电以及光热性能进行系统测试,和单一Zn基MOF相比,因引入三联吡啶Ru光敏单元,材料光吸收范围更广,光学带隙更窄。材料的光生载流子寿命更长,电荷转移电阻更低,载流子的分离与传输效率得到显著提升。在波长808 nm、功率0.8 W cm-2的激光照射下,样品仅用时1.3秒温度就迅速提升至155.7 ℃,光热转换速率十分突出,多次光照循环后,升温性能没有出现明显衰减,展现出良好的光热循环稳定性。
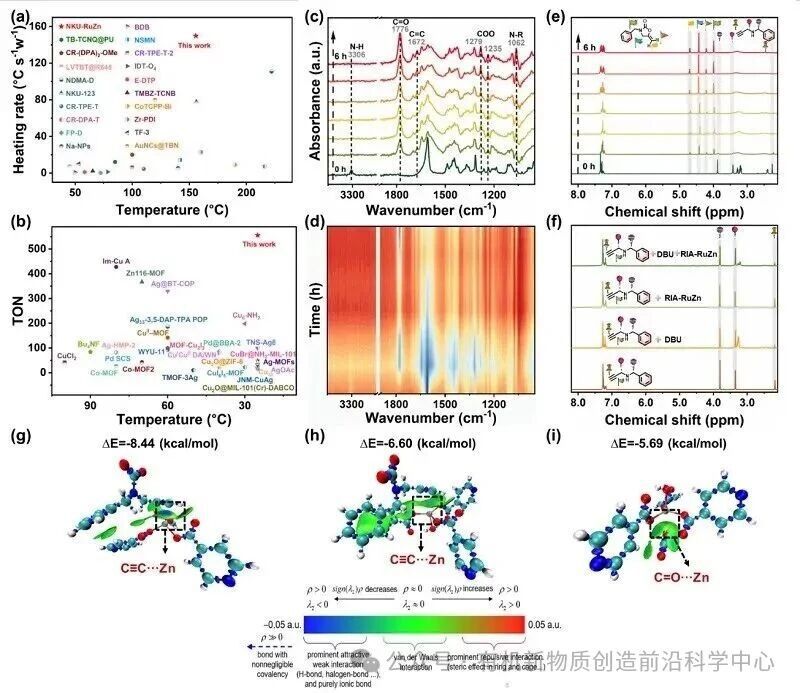
图4. GIA-RuZn光热、催化性能对比与
原位机理表征、理论吸附分析
实验选取炔丙胺与CO2的羧基环化反应作为催化评价体系,在20 W白光模拟太阳光的条件下开展催化测试。经过反应条件优化,以DBU为碱、乙腈为反应溶剂时,目标产物收率可以达到99%,转化数TON高达556。为了阐明GIA-RuZn催化活性的科学本质,研究团队通过原位谱学表征与密度泛函理论(DFT)计算对其反应路径进行了系统研究。在光照激发下,框架中的Ru单元发生电子跃迁,光生电子与空穴有效分离并迁移至活性位点。与此同时,骨架中的Zn催化位点与DBU碱分子协同作用,对炔丙胺底物的N-H键和末端炔烃进行活化,这在原位核磁共振In situ 1H NMR谱学上表现为终端炔烃质子信号的显著宽化与面积减小,从而为CO2的亲核进攻创造了有利条件。
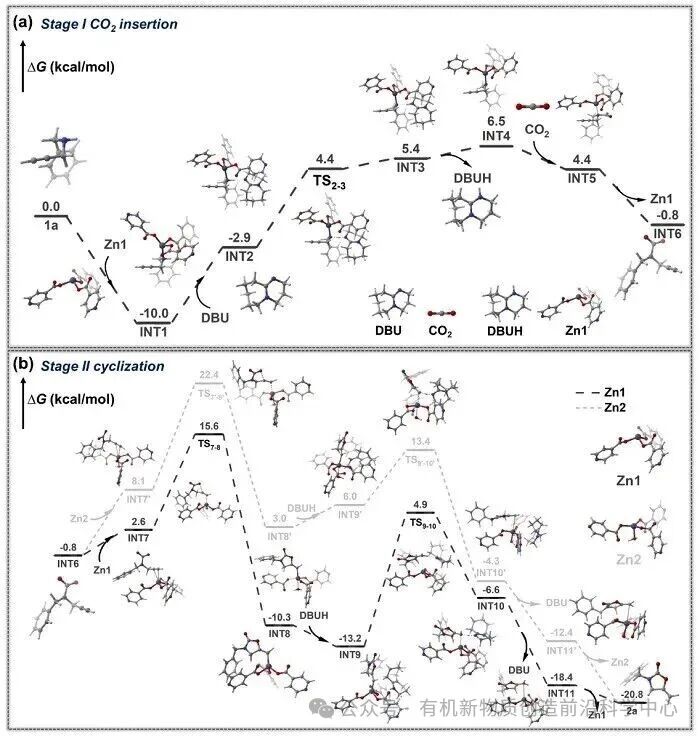
图5. DFT计算得到的GIA-RuZn催化下的反应自由能图
DFT 自由能计算表明,活化后的CO2分子进攻炔丙胺的氮原子以形成关键中间体 INT5,随后氮原子从锌位点上解离并产出中间体INT6,完成CO2插入阶段(Stage I)的反应。相比于缺乏锌中心协同配位的均相催化路径,GIA-RuZn 有效避免了高能垒中间体的产生,从而优化了热力学反应路径。在分子内环化与质子转移阶段(Stage II),MOF骨架独特的局域几何配位环境对稳定过渡态起到了决定性作用。计算对比表明,尽管体系中并存两种锌位点,但五配位的 Zn2位点由于配位趋于饱和,存在较大的空间位阻,导致其闭环环化能垒显著提高。相反,具有四面体环境的四配位Zn1位点在反应进程中显著稳定了环化过渡态(TS7-8),使该阶段反应能垒大幅降低。这种“光子驱动电荷转移”与“声子释放局域热能”在原子级尺度上的精密集成,使GIA-RuZn催化路径将全反应的整体自由能垒ΔG‡从无MOF参与条件下的29.1 kcal/mol显著拉低至18.1 kcal/mol。原位时间分辨红外(In situ FT-IR)光谱监测到的C=O、C=C等特征吸收峰随时间推进的动态演变,与理论计算结果实现了清晰的互证,在分子层面上解释了该材料在温和实验条件下具备高效CO2固定性能的原因。
03 总结
作者通过简便的一锅溶热法,成功构筑了一例新型异金属MOF(GIA-RuZn)。该材料具有独特的2D-to-3D交织穿透编织拓扑结构,不仅赋予了其优异的热稳定性,更使其在宽谱pH值(pH = 1-14)的水溶液中表现出优良的化学耐受性。光谱及同步辐射测试表明,光敏单元Ru(bpy)}3]2+与具催化活性的Zn2+位点在多孔单晶骨架内实现了原子级水平的精准集成与均匀分散。得益于微观热源与催化中心之间极近的空间临近性,GIA-RuZn 表现出显著的超快光热转换效应,在808 nm激光照射下仅需1.3 s表面温度便可由室温迅速升高至155.7 ℃。在20 W白光LED照射的温和条件下,GIA-RuZn 展现出卓越的CO2固定,催化炔丙胺与CO2的羧化环化反应目标产物产率高达99%,TON达到556,并可成功推进克级放大实验。原位实验与DFT理论计算表明,该材料通过“光子驱动电荷转移”与“声子释放局域热能”的原子级一体化协同,从根本上改变了反应活化路径,将全反应的整体自由能垒大大降低,为CO2固定的催化剂设计提供了良好的借鉴。
相关论文以“Ultrafast Photothermal Conversion of Ru/Zn Heterometallic Metal-Organic Framework for Synergistically Catalytic CO2 Fixation”为题发表于《Journal of the American Chemical Society》期刊,南开大学有机新物质创造前沿科学中心为论文通讯单位。
04 文献信息
Ultrafast Photothermal Conversion of Ru/Zn Heterometallic Metal-Organic Framework for Synergistically Catalytic CO2 Fixation. Fang-Yu Ren, Lu-Qiang Wang, Juan-Mei Wang, Zhi Fang, Ling-Hao Duan, Qi-Fan Wang, Min Zhou, Fang-Ting Zhu, Hong-Xia Shao, Qi Li, Shumei Su, Zhikang Cai, Donglin Wang, Yue Zhang, Hang Xu, Zhi Chen*, Ke R. Yang*, Bin Zhao*. J. Am. Chem. Soc., 2026, DOI: 10.1021/jacs.6c04262

关注“南开化学”微信公众号